





Oxidative stress and inflammation are determinant processes in the development of diabetic vascular complications. Heat shock protein 90 (HSP90) overexpression in atherosclerotic plaques plays a role in sustaining inflammatory mechanisms, and its specific inhibition prevents atherosclerosis. The present work investigates, in a mouse model of diabetes-driven atherosclerosis, whether atheroprotection by pharmacological HSP90 inhibition is accomplished by bolstering antioxidant defense mechanisms headed by nuclear factor erythroid-derived 2-like 2 (Nrf2).
MethodsStreptozotocin-induced diabetic apolipoprotein E-deficient mice were randomized to receive vehicle or HSP90 inhibitor (17-dimethylaminoethylamino-17-demethoxygeldanamycin, 4mg/kg) for 10 weeks. Aortic root sections were analyzed for plaque size and composition, transcription factor activity, and expression of inflammatory and antioxidant markers. In vitro studies were performed in murine macrophages cultured under hyperglycemic conditions.
ResultsTreatment with HSP90 inhibitor promoted the activation of Nrf2 in the aortic tissue of diabetic mice (predominantly localized in macrophages and smooth muscle cells) and also in cultured cells. Nrf2 induction was associated with a concomitant inhibition of nuclear factor-κB (NF-κB) in atherosclerotic plaques, thus resulting in a significant reduction in lesion size and inflammatory component (leukocytes and cytokines). Furthermore, atheroprotection by HSP90 inhibition was linked to the induction of cytoprotective HSP70, antioxidant enzymes (heme oxygenase-1, superoxide dismutase and catalase) and autophagy machinery (LC3 and p62/SQSTM1) in aortic tissue.
ConclusionHSP90 inhibition protects from atherosclerosis in experimental diabetes through the induction of Nrf2-dependent cytoprotective mechanisms, reinforcing its therapeutic potential.
Estrés oxidativo e inflamación son procesos clave en las complicaciones vasculares de la diabetes. La expresión de heat shock protein 90 (HSP90) en placas ateroscleróticas prolonga la inflamación, y su inhibición previene la aterosclerosis. Este trabajo investiga, en un modelo de aterosclerosis asociada a diabetes, si el efecto ateroprotector de la inhibición farmacológica de HSP90 está mediado por la inducción de mecanismos de defensa antioxidante dependientes del factor de transcripción nuclear factor erythroid-derived 2-like 2 (Nrf2).
MétodosRatones deficientes en apolipoproteína E con diabetes por estreptozotocina se trataron durante 10 semanas con el inhibidor de HSP90 (17-dimetilaminoetilamino-demetoxigeldanamicina, 4mg/kg). Analizamos tamaño/composición de las placas, factores de transcripción y marcadores inflamatorios y antioxidantes. In vitro estudiamos macrófagos murinos bajo condiciones de hiperglucemia.
ResultadosEl inhibidor de HSP90 indujo activación de Nrf2 en aorta de ratones diabéticos (localizado en macrófagos y células de músculo liso) y en células en cultivo. La activación de Nrf2 junto con la inhibición de nuclear factor-κB (NF-κB) en las placas ateroscleróticas resultó en una reducción significativa del tamaño de la lesión y del componente inflamatorio (leucocitos y citocinas). Este efecto ateroprotector se vinculó con un aumento de la proteína citoprotectora HSP70, enzimas antioxidantes (hemoxigenasa-1, superóxido dismutasa y catalasa) y genes de autofagia (LC3 y p62/SQSTM1) en aorta de ratones tratados con el inhibidor de HSP90.
ConclusiónLa inhibición de HSP90 protege del desarrollo de aterosclerosis en la diabetes experimental a través de la inducción de mecanismos citoprotectores dependientes de Nrf2, lo que refuerza su potencial terapéutico.
Diabetes is a common risk factor for the onset and progression of atherosclerosis. Consequently, cardiovascular disease has become the leading cause of mortality in diabetic patients.1 In addition to the strict control of metabolic derangements, there is an increasing need for strategies targeting diabetic macrovascular complications.2
Inflammation and oxidative stress play a key role in atherosclerosis particularly in the context of diabetes.3 Since they are tightly intertwined, it seems plausible that therapeutic strategies designed to counteract inflammation may also limit oxidative damage and vice versa. In this regard, we recently reported that the inhibition of heat shock protein 90 (HSP90), one of the most conserved chaperones in cellular stress response, interfered both the inflammatory responses4,5 and pro-oxidative factors6 in experimental atherosclerosis. HSP90 uses the energy generated by ATP binding and hydrolysis to ensure the proper conformation and functionality of its so-called “client” proteins, mostly involved in signal transduction and transcriptional regulation.7 To do so, HSP90 forms a multichaperone complex with other cochaperones, such as HSP70, that recognizes client proteins and modulates their activities.7 In this sense, HSP90 participates in the activation of nuclear factor-κB (NF-κB) pathway, crucial in cellular inflammation and immunity, by folding and activating the newly synthesized IκB kinase (IKK). An HSP90-stabilized IKK phosphorylates and induces proteasome degradation of the inhibitory protein IκBα.8 This allows NF-κB p65 subunit to enter the nucleus, promoting inflammatory response and pro-oxidant gene transcription. NF-κB can be inactivated by selective HSP90 inhibitors, such as geldanamycin and its derivatives, that block the ATP-binding site and promote both the degradation of IKK and the upregulation of the cytoprotective HSP70.9
Pharmacological and genetic studies revealed a functional crosstalk between NF-κB and nuclear factor erythroid-derived 2-like 2 (Nrf2),10 the main transcriptional regulator of cellular defense against oxidative stress and electrophiles. Upon oxidative stress, Nrf2 escapes its repressor Kelch-like ECH-associated protein 1 (Keap1), translocates into the nucleus and interacts with antioxidant response elements (ARE) to induce the expression of a battery of antioxidant enzymes, such as heme oxygenase-1 (HO-1), superoxide dismutase (SOD) and catalase.11 Evidence supports that NF-κB activity suppresses the transcriptional activity of Nrf2.10 However, this modulation is somewhat complex and possibly cell-dependent. NF-κB p65 subunit hampers Nrf2-ARE binding by helping the nuclear translocation of Keap112 or competing for transcriptional co-activators.13 Beyond this regulatory role, several protein interactors act as switchers between NF-κB and Nrf2 pathways.10
Because of the essential HSP90 action enabling NF-κB activation could also block Nrf2 as a result of the coordinated regulation existing between these two pathways,10 this work investigates whether targeting HSP90 activates Nrf2-mediated cytoprotection, thus contributing to the attenuation of diabetes-associated atherosclerosis. To that end, we assessed the effect of HSP90 inhibition on Nrf2 pathway in an experimental model of atherosclerosis combining hyperglycemia and hyperlipidemia.
Material and methodsDiabetes modelThe housing and care of animals and all the procedures performed in this study were strictly in accordance with the Directive 2010/63/EU of the European Parliament and were approved by the Institutional Animal Care and Use Committee of IIS-Fundacion Jimenez Diaz. Experimental diabetes was induced in male apolipoprotein E-deficient (apoE−/−) mice (Jackson Laboratory), at 8 weeks of age by intraperitoneal injection of streptozotocin (STZ, 125mg/kg/day (Sigma) in 10mM citrate buffer, pH 4.5) once a day for two consecutive days.5,14,15 Animals were maintained on standard diet and monitored every 2–3 days for body weight and non-fasting blood glucose. Mice with overt diabetes (glucose >19.4mmol/L) were randomized to receive 10 weeks of treatment with vehicle (200μL saline; n=8) or HSP90 inhibitor (17-dimethylaminoethylamino-17-demethoxygeldanamycin (InvivoGen), 4mg/kg; n=8) via intraperitoneal every second day. Age-matched apoE−/− mice were used as non-diabetic controls (vehicle; n=4). At the study endpoint, 16h-fasted mice were anesthetized (100mg/kg ketamine and 15mg/kg xylazine), saline-perfused and euthanized. Aortas were divided into two parts: the upper aortic root was embedded in OCT (Sakura Finetek) for histological analysis; the abdominal/thoracic aorta was processed for mRNA analysis. Serum concentrations of glucose and lipids were measured by automated methods.
Histological analysisAtherosclerotic lesions in serial 8μm thick sections (covering about 1000μm from valve leaflets) were quantified by morphometry after Oil Red O/hematoxylin staining. Individual lesion area was determined by averaging the maximal values (3–4 sections). Macrophages (MOMA-2, AbD Serotec; CD68, Abcam), T lymphocytes (CD3, DAKO), vascular smooth muscle cells (VSMC) (α-smooth muscle actin (SMA)-Cy3; Sigma), chemokine (C–C motif) ligand 2 (CCL2), tumor necrosis factor α (TNFα) (Santa Cruz Biotechnology) and HO-1 (Enzo Life Sciences) were detected by immunoperoxidase/immunofluorescence. Activated transcription factors were detected by in situ Southwestern histochemistry using digoxigenin-labeled probes.5,16 Positive staining was quantified using Image Pro-Plus (Media Cybernetics) and expressed as percentage or number of positive cells per lesion area.5,14
Cell culturesMurine bone marrow-derived macrophages were cultured for 7 days in DMEM with low glucose (5.5mmol/L) containing 10% FBS, 100U/mL penicillin, 100μg/mL streptomycin, 2mmol/L l-glutamine (Sigma), and 10% L929-cell conditioned medium as a source of macrophage colony stimulating factor.5,14 Quiescent cells were pretreated with HSP90 inhibitor (50nmol/L, 4h) before stimulation with high glucose (30mmol/L d-glucose (Sigma), 6h).
mRNA expression analysisTotal RNA from mouse aorta was extracted with TRIzol (Life Technologies). Target gene expression was analyzed by real-time quantitative PCR (Applied Biosystem) and normalized to 18S housekeeping gene.
Protein expression analysisTotal proteins from cultured cells were resolved on SDS–PAGE gels, transferred and immunoblotted for Ser40-phosphorylated-Nrf2 (P-Nrf2) (Bioss Antibodies) and α-tubulin (loading control; Sigma), using peroxidase conjugated secondary antibodies (Jackson ImmunoResearch). Blots were quantified using Quantity One software (Bio-Rad).
StatisticsValues are expressed as mean±SEM. Statistical analyses were performed using Prism 5 (GraphPad). Pearson's correlation analyses were performed for normally distributed parameters. Differences across groups were considered significant at p<0.05 using either non-parametric Mann–Whitney U test or one-way ANOVA followed by post hoc Bonferroni pairwise comparison test.
ResultsImpact of HSP90 inhibition on Nrf2 activation in diabetes-associated atherosclerosisIn order to explore the potential interplay between Nrf2 and HSP90 in the context of diabetic complications, we performed an experimental model of accelerated vascular injury with high similarities to human atherosclerosis, resulting from the combination of hyperglycemia and hyperlipidemia. In this study, STZ-induced diabetic apoE−/− mice were treated with either vehicle (control group) or HSP90 inhibitor (HSP90i group) for a period of 10 weeks.
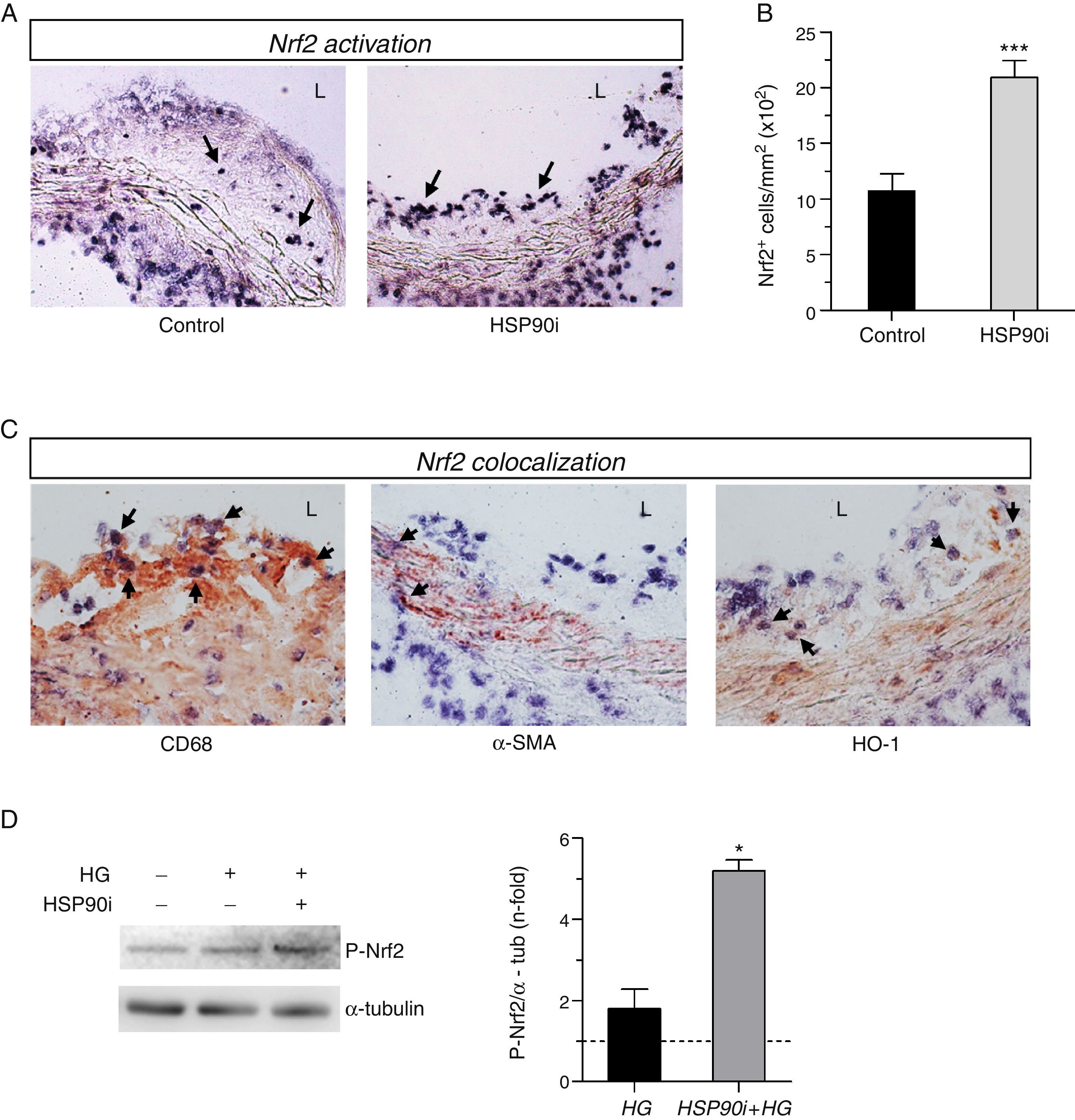
By using Southwestern histochemistry, a technique developed in our laboratory for in situ detection of activated transcription factors, we observed nuclear Nrf2 activation in aortic cells from intimal and medial layers of diabetic mice (Fig. 1A). Interestingly, quantification revealed a significant 2-fold increase of Nrf2-positive cells within the atherosclerotic lesions of mice receiving HSP90 inhibitor, compared with vehicle-treated mice (Fig. 1B). Double staining showed colocalization of activated Nrf2 with both CD68+ macrophages in the plaque and α-SMA+ VSMC in the media (Fig. 1C). Additionally, Nrf2 colocalize with HO-1 protein, a well-characterized Nrf2 target gene with a protective role against oxidative tissue injury (Fig. 1C). The impact of HSP90 inhibition on Nrf2 activation was also analyzed in vitro, in primary macrophages under high glucose conditions. Pretreatment of cells with HSP90 inhibitor increased the phosphorylation levels of Nrf2, thus confirming the in vivo findings (Fig. 1D).
 Representative images of in situ detection of activated Nrf2 in aortic sections of diabetic apoE−/− mice after 10 weeks of treatment with either vehicle (control) or HSP90 inhibitor (HSP90i). (B) Quantification of positive cells per lesion area. Mean±SEM of 8 animals per group. ***p<0.01 vs. control. (C) Colocalization of Nrf2 (blue-purple) with CD68, α-SMA, and HO-1 (red-brown immunoperoxidase). Magnifications ×200 (A) and ×400 (C); arrows indicate positive staining; L, lumen. (D) Western blot analysis of phosphorylated Nrf2 in bone marrow-derived macrophages pretreated with HSP90 inhibitor (50nmol/L, 4h) prior to stimulation with high glucose (HG, d-glucose 30mmol/L, 6h). Results expressed as relative increases over basal (horizontal dashed line) are mean±SEM of 3 independent experiments. *p<0.05 vs. basal.")
HSP90 inhibition activates Nrf2 in atherosclerotic lesions and in vitro. (A) Representative images of in situ detection of activated Nrf2 in aortic sections of diabetic apoE−/− mice after 10 weeks of treatment with either vehicle (control) or HSP90 inhibitor (HSP90i). (B) Quantification of positive cells per lesion area. Mean±SEM of 8 animals per group. ***p<0.01 vs. control. (C) Colocalization of Nrf2 (blue-purple) with CD68, α-SMA, and HO-1 (red-brown immunoperoxidase). Magnifications ×200 (A) and ×400 (C); arrows indicate positive staining; L, lumen. (D) Western blot analysis of phosphorylated Nrf2 in bone marrow-derived macrophages pretreated with HSP90 inhibitor (50nmol/L, 4h) prior to stimulation with high glucose (HG, d-glucose 30mmol/L, 6h). Results expressed as relative increases over basal (horizontal dashed line) are mean±SEM of 3 independent experiments. *p<0.05 vs. basal.
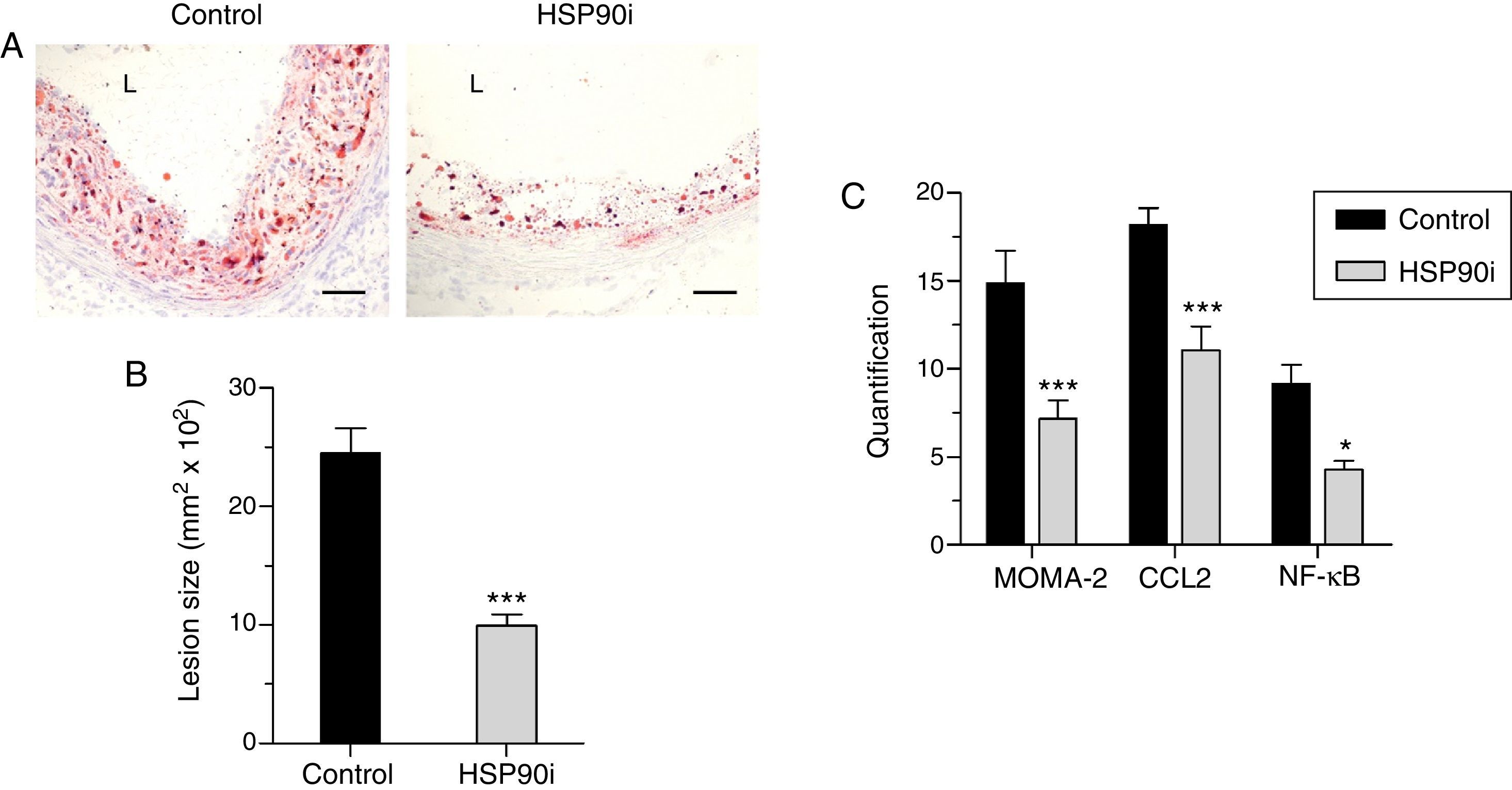
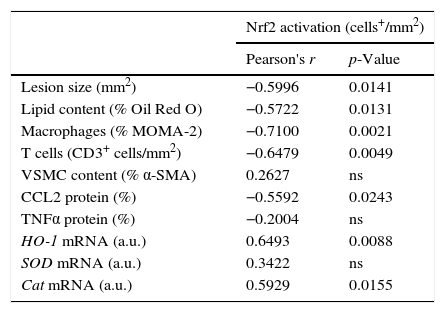
Quantitative assessment of atherosclerosis in aortic root sections revealed that therapy with HSP90 inhibitor reduced atherosclerotic lesion size in diabetic mice (% inhibition vs. control: 59±4, p<0.001) (Fig. 2A and B), an effect independent of changes in hyperglycemia and lipid profile (Table 1). Moreover, and consistent with our previous findings, plaques of HSP90 inhibitor-treated mice exhibited a less inflamed phenotype characterized by significant decreases in macrophage content, CCL2 expression, and NF-κB activation (% inhibition vs. control: 52±7, 39±7, and 53±6, respectively; Fig. 2C). Interestingly, the extent of Nrf2 activation in atherosclerotic plaques was inversely correlated with lesion size, lipid content, leukocyte numbers, CCL2 expression and NF-κB activation, but not with VSMC content and TNFα expression (Table 2 and Fig. 3A).
 Representative images (magnification ×200; L, lumen) of Oil Red O/hematoxylin staining in aortic root sections of diabetic apoE−/− mice treated with vehicle (control) and HSP90 inhibitor (HSP90i). (B) Quantification of lesion area. (C) Quantitative evaluation of inflammatory markers in atherosclerotic lesions. Results expressed as % of positive staining (MOMA-2 and CCL2; immunoperoxidase) or number of positive cells per lesion area (NF-κB; Southwestern histochemistry) are mean±SEM of 8 animals per group. *p<0.05, ***p<0.01 vs. control.")
Anti-atherosclerotic effect of HSP90 inhibition in diabetic mice: lesion size and inflammatory markers. (A) Representative images (magnification ×200; L, lumen) of Oil Red O/hematoxylin staining in aortic root sections of diabetic apoE−/− mice treated with vehicle (control) and HSP90 inhibitor (HSP90i). (B) Quantification of lesion area. (C) Quantitative evaluation of inflammatory markers in atherosclerotic lesions. Results expressed as % of positive staining (MOMA-2 and CCL2; immunoperoxidase) or number of positive cells per lesion area (NF-κB; Southwestern histochemistry) are mean±SEM of 8 animals per group. *p<0.05, ***p<0.01 vs. control.
Biochemical data of diabetic apoE−/− after 10 weeks of treatment.
| Vehicle (n=8) | HSP90 inhibitor (n=8) | |
|---|---|---|
| Glucose (mg/dL) | 516±7 | 526±15 |
| Total cholesterol (mg/dL) | 562±33 | 599±39 |
| HDL cholesterol (mg/dL) | 12±1 | 9±1 |
| LDL cholesterol (mg/dL) | 536±32 | 577±44 |
| Triglycerides (mg/dL) | 75±5 | 65±6 |
Data are expressed as mean±SEM of 8 animals per group. No significant differences were observed between the two groups.
Correlation between Nrf2 activation and atherosclerosis parameters in diabetic mice.
| Nrf2 activation (cells+/mm2) | ||
|---|---|---|
| Pearson's r | p-Value | |
| Lesion size (mm2) | −0.5996 | 0.0141 |
| Lipid content (% Oil Red O) | −0.5722 | 0.0131 |
| Macrophages (% MOMA-2) | −0.7100 | 0.0021 |
| T cells (CD3+ cells/mm2) | −0.6479 | 0.0049 |
| VSMC content (% α-SMA) | 0.2627 | ns |
| CCL2 protein (%) | −0.5592 | 0.0243 |
| TNFα protein (%) | −0.2004 | ns |
| HO-1 mRNA (a.u.) | 0.6493 | 0.0088 |
| SOD mRNA (a.u.) | 0.3422 | ns |
| Cat mRNA (a.u.) | 0.5929 | 0.0155 |
Pearson's correlation analysis in diabetic mice at the end of the study. ns, non-significant; a.u., arbitrary units.
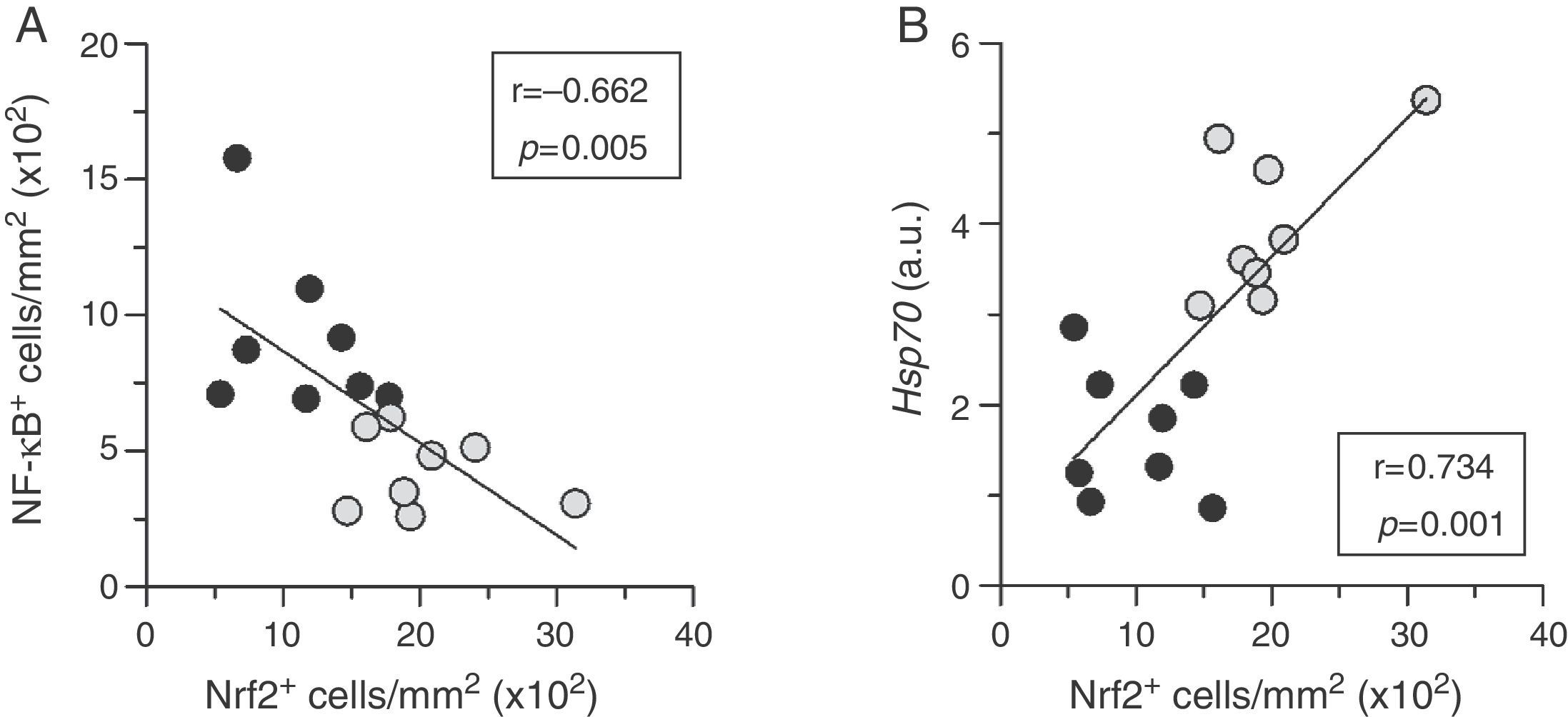
, and HSP70 mRNA expression (B) in atherosclerotic lesions from diabetic mice treated with either vehicle (black circles, n=8) or HSP90 inhibitor (gray circles, n=8). Pearson")
Nrf2 activation correlates to low inflammation and cytoprotection in atherosclerotic plaques. Correlation between the extent of activation of Nrf2 and NF-κB (A), and HSP70 mRNA expression (B) in atherosclerotic lesions from diabetic mice treated with either vehicle (black circles, n=8) or HSP90 inhibitor (gray circles, n=8). Pearson's r and p values are indicated.
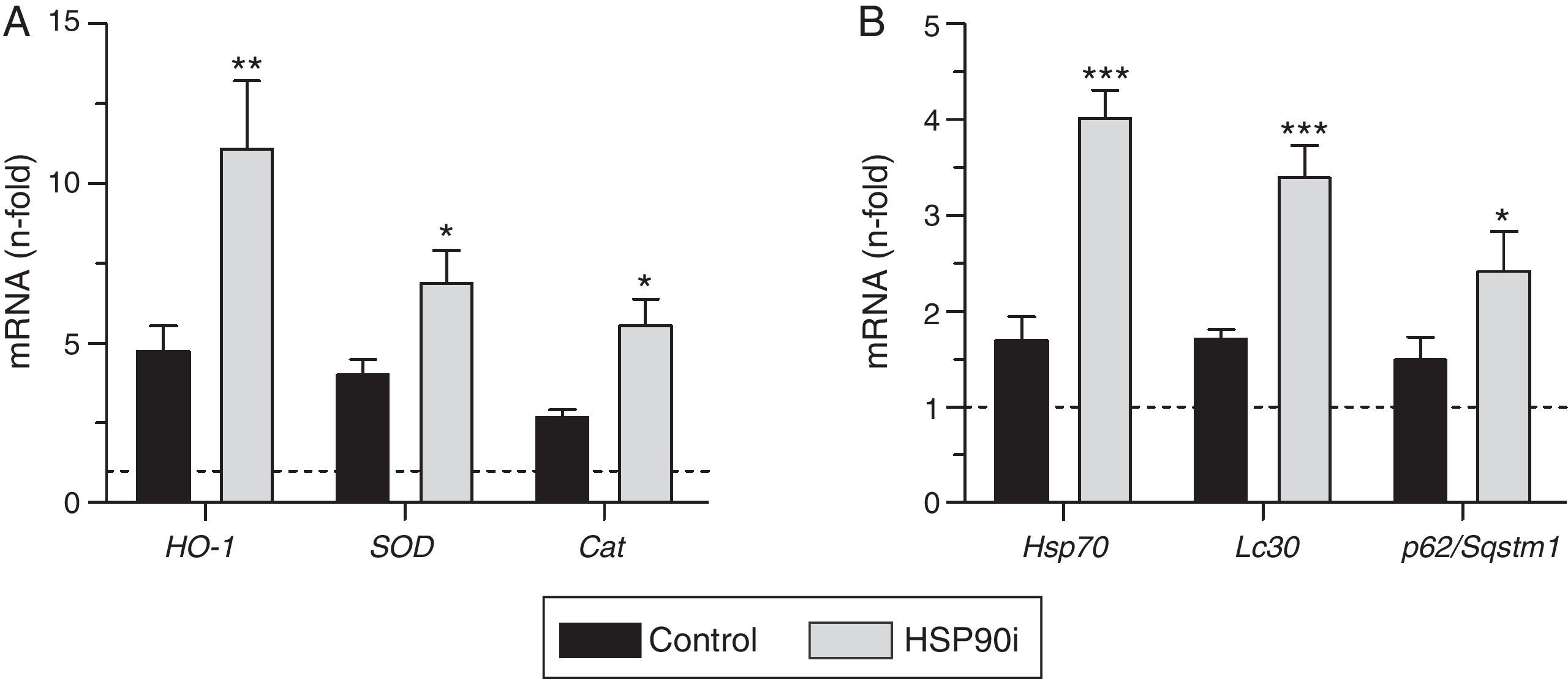
Next, we explored whether HSP90 inhibition affected the expression of antioxidant genes. Quantitative real-time PCR analysis on RNA from aorta revealed that the expression of HO-1, SOD, and catalase was increased by diabetes but further elevated after HSP90 inhibitor treatment (∼2-fold increase compared with vehicle-treated mice; Fig. 4A). Furthermore, the expression levels of HO-1 and catalase directly correlated with the extent of Nrf2 activation in atherosclerotic plaques (Table 2).
 (A), HSP90 cytoprotective cochaperone (Hsp70) and autophagy-involved genes (Lc3 and p62/Sqstm1), (B) in aortic tissue from diabetic mice treated with vehicle (control) and HSP90 inhibitor (HSP90i). Values normalized by 18S are expressed as fold increases vs. non-diabetic mice (horizontal dashed-lines). Data are the mean±SEM of 4–8 animals per group. *p<0.05, **p<0.01, ***p<0.01 vs. control.")
HSP90 inhibition triggers cytoprotective cellular mechanisms in the aorta of diabetic mice. Real-time PCR analysis of antioxidant defense genes (HO-1, SOD and Cat) (A), HSP90 cytoprotective cochaperone (Hsp70) and autophagy-involved genes (Lc3 and p62/Sqstm1), (B) in aortic tissue from diabetic mice treated with vehicle (control) and HSP90 inhibitor (HSP90i). Values normalized by 18S are expressed as fold increases vs. non-diabetic mice (horizontal dashed-lines). Data are the mean±SEM of 4–8 animals per group. *p<0.05, **p<0.01, ***p<0.01 vs. control.
Another cytoprotective effect of HSP90 inhibition resulting from the blockage of its ATP-binding sites is the upregulation of one of its cochaperones, including HSP70 protein.9 Consistent with this, we observed that treatment with HSP90 inhibitor increased the aortic mRNA expression of HSP70 (2.4-fold increase compared with vehicle-treated mice, p<0.001) (Fig. 4B), which directly correlated with Nrf2 activation levels (Fig. 3B).
Finally, we explored the effects of HSP90 inhibition on autophagy, an adaptative response that protects cells against injury, especially oxidative stress, by degrading damaged cellular material.17 Aortic tissue from diabetic mice treated with HSP90 inhibitor exhibited a significant increase in the expression levels of LC3 and p62/SQSTM1 (Fig. 4B), two key proteins participating in autophagy machinery.
DiscussionThe role of HSP90 is not just tied up to the heat shock response. Due to its chaperone condition, HSP90 takes part in multiple cellular processes and this fact turns it to a would-be target. This study raises an additional benefit of HSP90 targeting in diabetes-associated atherosclerosis, boosting the cytoprotection led by the evolutionary conserved Nrf2 pathway.
The present results show, for the first time, the in situ detection of Nrf2 activation in diabetic mouse aorta. Interestingly, we observed that HSP90 inhibition increased the number of Nrf2-activated cells (macrophages and VSMC) in atherosclerotic lesions and also promoted the phosphorylation/activation of Nrf2 in vitro. Despite being the master regulator of antioxidant defense, the role of Nrf2 in the pathogenesis of atherosclerosis is still controversial. In mice, total Nrf2 deficiency protects against diet-induced atherogenesis by affecting both systemic18,19 and local vascular mechanisms,20,21 whereas myeloid-specific Nrf2 deletion aggravates early and advanced stages of atherosclerosis.22,23 It has recently been reported that either natural or synthetic modulators of Nrf2 lessen diabetes-associated atherosclerosis in experimental models.24,25 In line with this, our results in diabetic mice revealed that HSP90 inhibition induced Nrf2 activation in aortic cells, and this was associated with a reduced size of atherosclerotic lesions and their lipid and inflammatory content, thus leading to a more stable plaque phenotype.
Concomitant with the induction of Nrf2 activity, we observed a reduced number of NF-κB-activated cells in atherosclerotic lesions of mice treated with HSP90 inhibitor, together with a down-regulation of chemokine expression and leukocyte infiltration. Although HSP90 is necessary to allow the relocalization and thus activation of the IKK in the NF-κB activation pathway,26 the dual effect of HSP90 inhibition on NF-κB and Nrf2 observed in diabetic mice is in line with the previously reported antagonistic effect of Nrf2 on the NF-κB pathway.10,27 Indeed, Nrf2 knockout mice showed enhanced NF-κB activity and increased expression of inflammatory genes,28–31 and this could be due to the modulation of IκBα degradation.31 Furthermore, Yu et al.12 identified a physical association between N-terminal region of activated NF-κB p65 subunit and Keap1 in the cytoplasm. This fact enabled p65 to assist Keap1 nuclear translocation, where blocked Nrf2-ARE-mediated transcriptional activity.12 Another well-established mechanism is based in the observations of Liu et al.32 that NF-κB p65 subunit repressed transcriptional activity of Nrf2-ARE system by sequestering nuclear coactivators, such as CREB-binding protein (CBP). Our studies with HSP90 inhibitor reinforce the potential therapeutic effect of the simultaneous modulation of NF-κB–Nrf2 axis in diabetic atherosclerosis, as previously reported in other inflammatory diseases.27,33
Limiting oxidative stress by bolstering antioxidant defense is a challenge to deal with diabetes-driven atherosclerosis. In this regard, the modulation of the antioxidant-mastering properties of Nrf2 has been assessed in diabetic vascular complications including nephropathy,24 retinopathy,34 and atherosclerosis.24,25 Accordingly, we analyzed the aortic expression of Nrf2-responsive antioxidant genes (HO-1, SOD, and catalase). HO-1 is a stress–response protein induced in atherosclerosis as a compensatory mechanism to reduce the oxidative injury in the arterial walls,35,36 whereas SOD and catalase work in concert to detoxify superoxide anions and hydrogen peroxide.37 In mice, both myeloid-specific and total deficiencies in HO-1 promoted atherosclerosis.38,39 Conversely, overexpression of all these antioxidant enzymes (HO-1, SOD and catalase) by either pharmacological or genetic manipulations prevented atherosclerosis progression.40–42 Interestingly, our data highlighted that diabetes promoted gene expression of these antioxidant enzymes in comparison with the non-diabetic controls. This fact reflected an adaptative response to the chronic exposure to oxidative stress associated with hyperglycemia, which has been previously reported in diabetes-sensitive tissues (i.e. kidney, retina, heart, or vasculature).2,43 Besides diabetes effect, we also found that HSP90 inhibition resulted in increased Nrf2 activity and subsequent induction of antioxidant Nrf2-dependent genes in the aortas of diabetic mice, thus reinforcing the HSP90 inhibition cytoprotective outcome on Nrf2-mediated antioxidant defense.
An established protective effect of HSP90 inhibition is the induction of HSP70, as a result of the activation its transcriptional regulator.9 In line with our previous studies pointing out the anti-inflammatory effect of HSP70 in atherosclerosis,4,5 the present work unveils a direct association between HSP70 upregulation and Nrf2 activation, both resulting from HSP90 inhibition, in the atherosclerotic lesions of diabetic mice.
Our observations in diabetic mice revealed that HSP90 inhibition could also participate in autophagic machinery. Autophagy is a reparative, life-sustaining adaptative response by which damaged cellular components are trapped in double-membrane vesicles (called autophagosomes) and degraded upon fusion with lysosomes, forming autophagolysosomes.44 LC3 and p62/SQSTM1 are considered useful protein markers of autophagic status.45 Autophagy is triggered by inflammation and oxidative stress during the atherosclerotic process.46,47 However, up to a point where cellular damage in atherosclerotic plaques becomes so overwhelming that autophagic machinery is not able to cope with it, and autophagic flux ends blocked.17 Razani et al.48 showed that p62/SQSTM1 was dramatically increased in atherosclerotic aortas of high fat-fed apoE−/− mice. Our results are apparently contradictory with a beneficial effect of HSP90 inhibition on autophagy, due to the fact that the treatment upregulated both LC3 and p62/SQSTM1 in the aortas of diabetic mice. Nevertheless, we propose that this increase in gene expression could mean a certain restoration of autophagy in the aorta of diabetic mice, which was doubly jeopardized by atherosclerosis and hyperglycemic conditions. LC3 undergoes transcriptional and posttranscriptional regulation, so the assessment of mRNA levels helps to interpret autophagic status.45 Interestingly, our results pointed out that the induction of p62/SQSTM1 expression could be related to the enhanced aortic Nrf2 activation resulting from HSP90 inhibition. This observation is in line with the recently described crosstalk between the Nrf2-Keap1-ARE axis and autophagy, where p62/SQSTM1 leads a non-canonical Nrf2 activation pathway through the physical sequestration and turnover of Keap1.49,50
It is worth to state a limitation of our study. Due to the lack of aortic protein samples, we only presented data from analysis on gene expression at mRNA level. However, protein expression data should be more adequate to assess the effectiveness of HSP90 inhibition on autophagy. More studies are needed to fully characterize this effect.
Taken together, our data strengthen the pharmacotherapeutic benefits of HSP90 inhibition in the protection against diabetes-driven atherosclerosis, by uncovering its effects on Nrf2 pathway, antioxidant defense and autophagy-mediated cytoprotection.
FundingThis work was supported by funds from the Sociedad Española de Arteriosclerosis (Basic Research Award 2012), MINECO (SAF2012-38830, SAF2015-63696-R) and Instituto de Salud Carlos III (FIS/FEDER PI14/00386, PIE13/00051). IL was supported by a postdoctoral fellowship from FIS (Sara Borrell program).
Author contributionsIL and CGG designed the study, researched and analyzed data, and wrote the manuscript. AO and CR researched, analyzed data and critically revised the manuscript. LLS and SB evaluated in vivo and in vitro data. JE reviewed the manuscript for intellectual content.
Conflict of interestNo potential conflicts of interest relevant to this article were reported.
The authors greatly acknowledge the initial contribution of Dr. B Mallavia (Renal, Vascular and Diabetes Research Lab, IIS-Fundacion Jimenez Diaz) to the animal models. The authors also thank Dr. J Madrigal-Matute (Dept. of Developmental and Molecular Biology, Albert Einstein College of Medicine, New York) and Dr. JL Martin-Ventura (Renal, Vascular and Diabetes Research Lab, IIS-Fundacion Jimenez Diaz) for their helpful guidance on autophagy and HSP90 fields.