




NF-kB. Conclusions. These findings indicate that negative cross-talk between NF-*B and PPAR may interfere in the transactivation capacity of the latter, leading to a fall in the expression of genes involved in fatty acid metabolism, and that these changes are prevented by atorvastatin.
Introducción
El desarrollo de la hipertrofia cardíaca se considera una respuesta fisiológica adaptativa a una gran variedad de estímulos extrínsecos, como la hipertensión arterial, la enfermedad cardíaca valvular, el infarto de miocardio y la miocardiopatía. Aunque inicialmente el desarrollo de este proceso puede ser beneficioso, al normalizar el estrés sobre la pared y preservar una función cardíaca normal, su mantenimiento durante tiempo prolongado es una de las principales causas de insuficiencia cardíaca, arritmia y muerte súbita1,2. Además, la hipertrofia cardíaca es un factor de riesgo independiente de enfermedad cardiovascular, ya que incrementa la mortalidad cardiovascular más de 2 veces1,3.
Entre las vías de señalización implicadas en el crecimiento hipertrófico del miocardio, la vía del factor nuclear (NF)-kB desempeña un papel clave. Se ha demostrado que la inhibición de la actividad NF-kB bloquea o atenúa la respuesta hipertrófica de los miocitos cardíacos en cultivo4-7. La forma activada de NF-kB es un heterodímero, habitualmente integrado por 2 proteínas, las subunidades p65 y p50. En las células no estimuladas, NF-*B se encuentra en el citoplasma unido a IkB, una proteína inhibidora que evita su entrada en el núcleo. Sin embargo, cuando las células son estimuladas, unas cinasas específicas fosforilan IkB y provocan su degradación en el proteasoma. La liberación de IkB permite al heterodímero NF-kB alcanzar el núcleo, donde se une a secuencias específicas en el promotor de los genes diana de este factor de transcripción. A través de este mecanismo, NF-kB desempeña un papel fundamental en el control de las respuestas inmunes e inflamatorias.
Por otro lado, el desarrollo de la hipertrofia cardíaca se asocia con un aumento en la utilización de glucosa y un descenso en la oxidación de ácidos grasos, una característica del corazón fetal8,9. La expresión de los genes cardíacos implicados en el metabolismo de la glucosa y los ácidos grasos se controla por los receptores activados por proliferadores peroxisómicos (peroxisome proliferator-activated receptors [PPAR]). La subfamilia PPAR está formada por 3 miembros, PPARα (NR1C1 según la nomenclatura unificada para la superfamilia de los receptores nucleares), PPARβ/δ (NR1C2) y PPARγ (NR1C3). PPARα se expresa principalmente en tejidos que poseen una elevada capacidad de catabolizar ácidos grasos, como el hígado, el riñón, el corazón y el músculo esquelético10; PPARβ/δ se expresa en la mayoría de los tejidos, y PPARγ presenta un patrón de expresión más restringido, expresándose principalmente en el tejido adiposo, mientras que otros tejidos, como el músculo esquelético y el corazón, contienen cantidades limitadas. Es interesante destacar que se ha descrito una interrelación negativa entre PPARα y NF-kB11,12.
En los últimos años se ha sugerido que los PPAR pueden desempeñar un papel muy importante en la enfermedad cardíaca. Por ejemplo, se ha descrito que el desplazamiento en la utilización de sustrato desde los ácidos grasos hacia la glucosa que se produce durante el desarrollo de la hipertrofia cardíaca se asocia con una desactivación de PPARα13. Estos datos sugieren que una reducción de la actividad de este factor de transcripción puede estar implicada en la disminución de las enzimas implicadas en la oxidación de los ácidos grasos. Sin embargo, el papel de PPARβ/δ en el desarrollo de este proceso sigue siendo desconocido. Recientemente, Gilde et al14, mediante la utilización de cardiomiocitos de rata neonatales y de células H9c2 derivadas de corazones embrionarios de rata, demostraron que PPARβ/δ es el tipo de PPAR predominante en las células cardíacas y que desempeña un papel muy importante en la regulación del metabolismo lipídico cardíaco. Estos datos sugerían que, de forma similar a PPARα, PPARβ/δ podía ser muy importante en la enfermedad cardíaca.
Los tratamientos actuales de la hipertrofia cardíaca se limitan a vasodilatadores o reductores de poscarga, con pocos fármacos dirigidos a los procesos cardíacos. Los inhibidores de la 3-hidroxi-3-metilglutaril-CoA (HMG CoA) reductasa, o estatinas, se prescriben fundamentalmente como agentes hipocolesterolemiantes que reducen la incidencia de infartos de miocardio y los accidentes cerebrovasculares. Además de inhibir la síntesis de colesterol, las estatinas también inhiben la síntesis de importantes intermediarios isoprenoides que son fundamentales para el anclaje a la membrana plasmática y la correcta funcionalidad de una gran variedad de proteínas. Además, se ha citado recientemente que las estatinas previenen la hipertrofia cardíaca por su capacidad para inhibir la generación de especies reactivas del oxígeno15. Sin embargo, se desconoce todavía si durante este proceso las estatinas previenen la activación de NF-kB y si este efecto puede estar relacionado con los cambios en el metabolismo de los ácidos grasos. En este estudio, hemos examinado el papel de la atorvastatina sobre la actividad NF-kB y PPAR en la hipertrofia cardíaca por sobrecarga de presión.
Material y método
Material
La atorvastatina fue proporcionada por los laboratorios Pfizer. El [γ-32P] dATP (3000 Ci/mmol) se obtuvo de Amersham Pharmacia Biotech KK, y los demás reactivos, de Sigma.
Preparación y análisis del ARN
El ARN total se aisló mediante el reactivo Ultraspec (Biotecx, Houston). El ARN total aislado por este medio no está degradado y está libre de contaminación por proteínas o ADN. La cuantificación de los valores relativos de ARNm se determinó por la técnica de transcriptasa reversa unida a la reacción en cadena de la polimerasa (RT-PCR) tal como se describió previamente16. Las secuencias sentido y antisentido de los oligonucleótidos utilizados para la amplificación fueron los siguientes: factor atrial natriurético (atrial natriuretic factor [ANF]), 5'-TCCTCTTCCTGGCCTTTTGGC-3' y 5'-AGACGGGTT GCTTCCCCAGTC-3'; α-actinina, 5'-GGCTGTGTTCCCATCCAT-CGT-3' y 5'-CCCGGTTAGCTTTGGGGTTCA-3', piruvatodeshidrogenasa cinasa 4 (PDK4), 5'-GAACACCCCTTCCGTCCA GCT-3' y 5'-TGTGCCATCGTAGGGACCACA-3' y APRT (adenosil fosforribosil transferasa), 5'-GCCTCTTGGCCAGTCACCTGA-3' y 5'-CCAGGCTCACACACTCCACCA-3'. La amplificación de cada gen proporcionó una única banda del tamaño esperado (ANF: 234 pares de bases [pb], α-actinina: 266 pb y APRT: 329 pb). Los resultados de la expresión específica de cada ARNm se expresan siempre respecto a la expresión del gen control (APRT).
Hipertrofia cardíaca inducida por sobrecarga de presión
Ratas macho Sprague-Dawley (225 a 250 g) mantenidas en condiciones estándar de iluminación (ciclo de luz-oscuridad de 12 h) y temperatura (21 ± 1 ºC) fueron alimentadas con una dieta estándar (Panlab, Barcelona) durante 5 días antes de iniciar el estudio. Los animales se distribuyeron aleatoriamente en 3 grupos: a) ratas control operadas pero en las que no se provocó la hipertrofia cardíaca (sham-operated); b) ratas sometidas a constricción de la aorta (banded rats), y c) ratas sometidas a constricción de la aorta tratadas con atorvastatina. Cinco días antes del procedimiento quirúrgico, las ratas se alimentaron con una dieta control o una dieta con 0,05% (p/p) de atorvastatina (que proporcionaba unos 15 mg/kg/día de atorvastatina aproximadamente). Las dietas se prepararon tal como se describió en un estudio previo17. A lo largo del tratamiento se determinó el peso y la ingesta de dieta. La sobrecarga de presión fue inducida por constricción de la aorta abdominal a escala suprarrenal mediante una seda de nylon 7-0 atada a una aguja despuntada (25 G, diámetro interno de 0,625 mm) para conseguir el mismo diámetro y que posteriormente fue retirada. En las ratas operadas pero no sometidas a sobrecarga de presión, se realizó el mismo procedimiento excepto que la sutura no se ató alrededor de la aorta. Los corazones se extrajeron 15 días después del procedimiento quirúrgico. Posteriormente se determinó la relación entre peso corazón/peso corporal (HW/BW) y las muestras fueron congeladas en nitrógeno líquido a -80 ºC.
Inmunodetección
Los lisados celulares y los extractos nucleares de corazón se obtuvieron tal como se describió previamente16. Las proteínas (30 µg) se separaron en geles de electroforesis de poliacrilamida y se transfirieron a membranas Immobilon de difluoruro de polivinilideno (Millipore, Bedford, MA). El análisis de las proteínas se realizó utilizando anticuerpos contra IkBα, IkBβ, p65 (Santa Cruz Biotechnology) y β-tubulina (Sigma). La detección se realizó utilizando el kit de quimioluminiscencia EZ-ECL (Biological Industries, Beit Haemek Ltd., Israel). El tamaño de las proteínas detectadas se estimó utilizando estándares de peso molecular (Life Technologies).
Ensayos de retardación de la movilidad electroforética
El aislamiento de los extractos nucleares se realizó tal como se describió previamente16. Los ensayos de retardación de la movilidad electroforética (electrophoretic mobility shift assay [EMSA]) se realizaron utilizando oligonucleótidos de doble cadena (Promega, Madison, WI) para el lugar de unión consenso de NF-kB (5'AGTTGAGGGGACTTTCCCAGGC-3') y Oct-1 (5'-TGTCGAATGCAAATCACTAGAA-3'). Los oligonucleótidos se marcaron en la siguiente reacción: 2 µl de oligonucleótido (1,75 pmol/µl), 2 µl de 5x tampón cinasa, 1 µl de T4 polinucleótido quinasa (10 U./µl), y 2,5 µl de [*-32P] ATP (3.000 Ci/mmol a 10 mCi/ml) incubados a 37 ºC durante 1 h. La reacción se paró mediante la adición de 90 µl de tampón TE (10 mM Tris-HCl pH 7,4 and 1 mM EDTA). Para separar la sonda marcada del ATP no unido, la mezcla de la reacción se eluyó en una columna Nick (Pharmacia, Sant Cugat, Barcelona, Spain), según las instrucciones del fabricante. Las proteínas nucleares (10 µg) se incubaron durante 10 min en un baño de hielo en tampón de unión (10 mM Tris-HCl pH 8,0, 25 mM KCl, 0,5 mM DTT, 0,1 mM EDTA pH 8,0, 5% glicerol, 5 mg/ml BSA, 100 µg/ml ARNt y 50 µg/ml poli [dI-dC]), en un volumen final de 15 µl. La sonda marcada (aproximadamente, 60.000 cpm) se añadió a la reacción y se incubó durante 15 min a 4 ºC. Donde se indica, se añadió un oligonucleótido competidor específico antes que la sonda marcada y se incubó durante 10 min en un baño de hielo. El anticuerpo contra p65 se añadió 15 min antes de la incubación con la sonda marcada a 4 ºC. Los complejos proteína-ADN se separaron mediante electroforesis a 4 ºC en un gel de poliacrilamida al 5% y se sometieron a autorradiografía.
Coinmunoprecipitación
Los extractos nucleares celulares se llevaron a un volumen final de 0,5 ml con tampón que contenía 10 mM PBS, 50 mM KCl, 0,05 mM EDTA, 2,5 mM MgCl2, 8,5% glicerol, 1 mM ditiotreitol, 0,1% Triton X-100, BSA 2% y 1 mg/ml de leche desnatada durante 6 h a 4 ºC, y se incubaron con 4 µg de anti-p65. Los inmunocomplejos se capturaron incubando las muestras en una suspensión de proteína A-agarosa durante toda la noche a 4 ºC en un agitador orbital. Las bolas de agarosa se recogieron por centrifugación y se lavaron 3 veces con PBS que contenían inhibidores de las proteasas. Tras la centrifugación, el precipitado se lavó con 60 µl de tampón de muestra y se llevó a ebullición durante 5 min a 100 ºC. Una alícuota del sobrenadante se sometió a electroforesis en un gel de acrilamida al 10% y se incubó con anticuerpos contra PPARα o β/δ.
Análisis estadístico
Los resultados corresponden a un mínimo de 4 experimentos independientes. Las diferencias significativas se establecieron por la t de Student o por ANOVA de una vía, según el número de grupos comparados, con la utilización del programa GraphPad Instat V2.03 (GraphPad Softwware Inc., San Diego, CA). Las diferencias se consideraron significativas cuando p < 0,05.
Resultados
El tratamiento con atorvastatina previene la aparición de hipertrofia cardíaca inducida por sobrecarga de presión
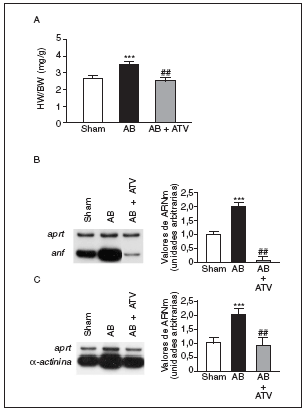
La hipertrofia cardíaca se caracteriza por un incremento en la relación entre el peso del corazón y el peso corporal (HW/BW), la inducción de genes fetales (como por ejemplo ANF) y una desorganización del sarcómero. Por ello, inicialmente determinamos los efectos del tratamiento con atorvastatina sobre estos parámetros en un modelo animal de hipertrofia cardíaca inducida por sobrecarga de presión. La relación HW/BW aumentó significativamente (1,35-veces; p < 0,001) en las ratas sometidas a constricción de la aorta comparadas con las ratas control (sham-operated rats) (fig. 1a). El tratamiento con atorvastatina eliminó completamente el aumento en la relación HW/BW (p < 0,01 comparado con las ratas sometidas a constricción de la aorta, pero no tratadas). Además, la sobrecarga de presión también causó un aumento de 2 veces en los valores de ARNm de ANF comparado con las ratas control (fig. 1b). Sin embargo, en el corazón de las ratas tratadas con atorvastatina la expresión de ANF casi desapareció. De forma parecida, los valores de ARNm de la proteína asociada al sarcómero α-actinina aumentaron 2 veces comparadas con las ratas control (fig. 1c), mientras que en el corazón de las ratas tratadas con atorvastatina esta inducción no se observó.
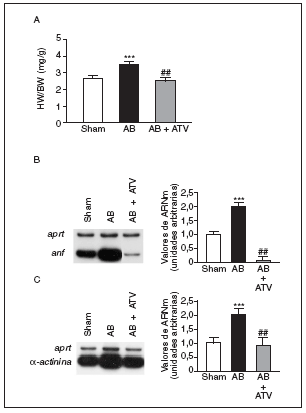
Figura 1. La atorvastatina inhibe la hipertrofia cardíaca inducida por sobrecarga de presión. La inducción de la sobrecarga de presión se llevó a cabo mediante constricción de la aorta abdominal. El tratamiento con atorvastatina se inició 5 días antes del procedimiento quirúrgico y continuó durante 15 días. El fármaco se administró mezclado con la dieta a una concentración del 0,05% (p/p). Pasados 15 días desde la operación, los corazones se diseccionaron y pesaron. A) Análisis de la relación peso corazón/peso corporal (HW/BW) en ratas control operadas pero sin constricción de la aorta (sham-operated rats), en ratas con constricción de la aorta (aortic banding [AB]) y en ratas con constricción de la aorta tratadas con atorvastatina (AB + ATV). Análisis de los valores de ARNm de la ANF (B) y la α-actinina (C) en la hipertrofia cardíaca por sobrecarga de presión. Se presenta una autorradiografía representativa. Los datos se expresan como la media ± desviación estándar de 4 experimentos diferentes. *p < 0,001 respecto a las ratas control. ##p < 0,01 respecto a las ratas con constricción de la aorta.
La atorvastatina previene la reducción de la expresión de PPARα y β/δ en la hipertrofia cardíaca inducida por sobrecarga de presión
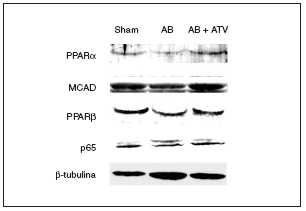
Se ha demostrado que la inducción de hipertrofia cardíaca provoca una caída en la expresión de los valores de ARNm de PPARα y de varios de sus genes diana implicados en el metabolismo de los ácidos grasos13. Sin embargo, se desconocen los efectos de la hipertrofia cardíaca sobre la expresión de PPARβ/δ. Por esta razón, se determinaron las consecuencias de la hipertrofia cardíaca inducida por sobrecarga de presión y del tratamiento con atorvastatina sobre la expresión proteica de estos factores de transcripción. Tal como se muestra en la figura 2, se observó una caída en la expresión de proteína PPARα (el 60% de reducción; p < 0,05) en los corazones de las ratas sometidas a constricción de la aorta comparada con las ratas control. Además, los valores de proteína de la deshidrogenasa de acil-CoAs de cadena media (MCAD), un gen diana de PPARα, también disminuyeron un 40% (p < 0,05). Hay que destacar que el tratamiento con atorvastatina previno la reducción de la expresión tanto de PPARα como de la MCAD. Igualmente, la presencia de hipertrofia cardíaca causó una reducción de la expresión de la proteína PPARβ/δ del 50% (p < 0,05), lo que indica que el patrón de expresión de PPARα y β/δ durante la hipertrofia cardíaca presenta cambios similares. El tratamiento con atorvastatina también evitó la reducción de PPARβ/δ durante la hipertrofia cardíaca. Finalmente, la expresión de la subunidad p65 de NF-kB no se modificó ni por la constricción de la aorta ni por el tratamiento con atorvastatina.
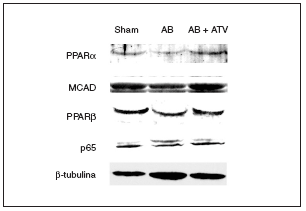
Figura 2. El tratamiento con atorvastatina previene la reducción de los valores de proteína de los PPAR y la MCAD en la hipertrofia cardíaca por sobrecarga de presión. Los extractos proteicos cardíacos de las ratas control operadas (sham-operated rats), las ratas con constricción de la aorta (aortic banding [AB]) y las ratas con constricción de la aorta tratadas con atorvastatina (AB + ATV) se analizaron por inmunodetección con anticuerpos contra PPARα, MCAD, PPARβ/δ, p65 y β-tubulina.
El tratamiento con atorvastatina previene la activación de NF-kB en la hipertrofia cardíaca inducida por sobrecarga de presión
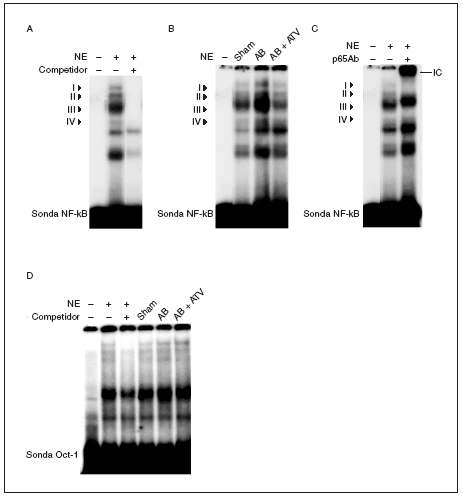
Puesto que la activación de NF-kB es necesaria para el crecimiento hipertrófico de los cardiomiocitos18-21 y se ha demostrado que las estatinas inhiben la activación de este factor de transcripción en células vasculares22, realizamos ensayos de retardación de la movilidad electroforética (EMSA) para investigar si la atorvastatina inhibía la activación de NF-kB en la hipertrofia cardíaca inducida por sobrecarga de presión. Estos estudios demostraron que la sonda NF-kB formaba 4 complejos específicos con las proteínas cardíacas nucleares (complejos I a IV; fig. 3a), según los experimentos de competición realizados mediante la adición de un exceso de oligonucleótido NF-kB no marcado a las mezclas de incubación. La actividad de unión NF-kB aumentó en las ratas sometidas a constricción de la aorta, especialmente el complejo III, comparado con las ratas control (fig. 3b) y este efecto se abolió por el tratamiento con atorvastatina. La caracterización de NF-kB se realizó incubando los extractos nucleares con un anticuerpo dirigido contra la subunidad p65 de este factor de transcripción. La adición de este anticuerpo a las mezclas de incubación provocó un retardamiento adicional del complejo III, que demostró que estaba formado por p65 (fig. 3c). No se observaron cambios en la unión al ADN de las proteínas cardíacas nucleares a una sonda Oct-1 en los diferentes grupos de ratas, lo que demostraba que el aumento observado para la sonda NF-*B era específico (fig. 3d). En conjunto, estos datos demuestran que la atorvastatina inhibe la activación de NF-kB en la hipertrofia cardíaca por sobrecarga de presión y que este mecanismo puede contribuir al efecto antihipertrófico de este compuesto.
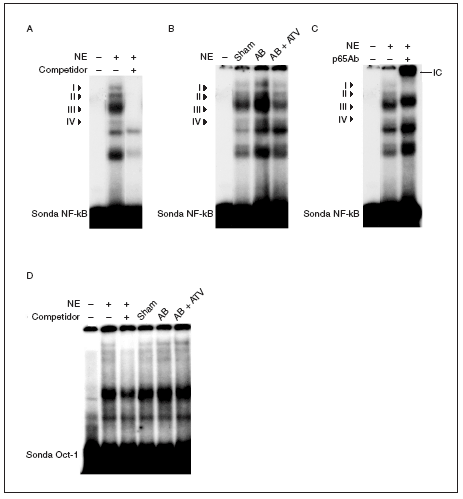
Figura 3. El tratamiento con atorvastatina previene la activación de NF-kB en la hipertrofia cardíaca inducida por sobrecarga de presión. A) Autorradiografía del ensayo de EMSA realizado con el oligonucleótido NF-kB marcado con 32P y extracto nuclear de proteínas (nuclear extract [NE]) que muestra 4 complejos específicos (I a IV), según los resultados de la competición con un exceso molar de sonda no marcada. B) Autorradiografía del EMSA realizado con el oligonucleótido NF-kB marcado con 32P y NE cardíacos de ratas control operadas (sham-operated rats), ratas con constricción de la aorta (aortic banding [AB]) y ratas con constricción de la aorta tratadas con atorvastatina (AB + ATV). C) Análisis de superretardación realizado mediante incubación de NE con un anticuerpo contra la subunidad p65 de NF-kB. Se ha marcado el inmunocomplejo superretardado (supershifted immune complex [IC]) que se forma. D) Autorradiografía del ensayo de EMSA realizado con el oligonucleótido Oct-1 marcado con 32P.
El tratamiento con atorvastatina inhibe la interacción p65-PPAR en la hipertrofia cardíaca inducida por sobrecarga de presión
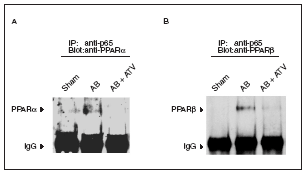
La desactivación de PPARα durante la hipertrofia cardíaca se ha relacionado con la fosforilación de esta proteína13. En este trabajo intentamos descubrir si existen otros mecanismos que pueden contribuir a reducir la vía de señalización de los PPAR. Puesto que se ha citado la existencia de una interacción física entre PPARα y la subunidad p65 de NF-kB11,12, y que NF-kB inhibe la expresión de los genes diana de PPARα23, seguidamente evaluamos si la presencia de hipertrofia cardíaca se traducía en un aumento de la interacción p65-PPAR. Para ello los extractos nucleares aislados de los corazones se inmunoprecipitaron con la utilización de un anticuerpo contra p65 acoplado a partículas de proteína A-agarosa. Los inmunoprecipitados se sometieron a electroforesis de poliacrilamida y se inmunodetectaron con anticuerpos anti-PPARα y anti-PPARβ/α. Los datos presentados en la figura 4 demuestran que la inducción de hipertrofia cardíaca por sobrecarga de presión aumentó la interacción física de p65 con PPARα y β/α, lo que sugiere que al aumento de esta asociación es un mecanismo que contribuye a la reducción de la expresión de los genes diana de PPAR implicados en el metabolismo de los ácidos grasos. Es importante destacar que en los corazones de las ratas tratadas con atorvastatina no se observó aumento de la interacción física entre p65 y PPAR, lo que sugiere que prevenir la activación de NF-kB evita esta interacción.
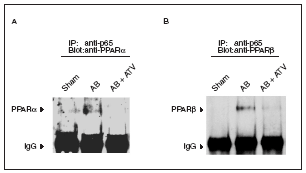
Figura 4. La atorvastatina reduce la asociación de los PPAR con la subunidad p65 de NF-kB en la hipertrofia cardíaca inducida por sobrecarga de presión. Los extractos nucleares (igualados según la concentración de proteína) de ratas control operadas (sham-operated rats), ratas con constricción de la aorta (aortic banding [AB]) y ratas con constricción de la aorta tratadas con atorvastatina (AB + ATV) se sometieron a inmunoprecipitación con la utilización de anticuerpos anti-p65 acoplados a partículas de proteína A-agarosa. Los inmunoprecipitados se sometieron a inmunodetección con anticuerpos anti-PPARα (A) y anti-PPARβ/α (B). Las flechas representan las señales correspondientes al tipo de PPAR o a las IgG.
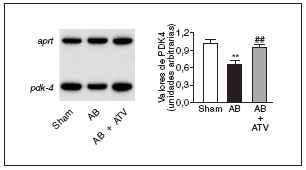
Finalmente, para determinar si la prevención de la actividad NF-kB por atorvastatina mejoraba la expresión de genes diana de PPAR implicados en el metabolismo de los ácidos grasos, se valoraron los valores de ARNm de la PDK4, un gen diana de PPAR, tanto α como β/δ14, que suprime la oxidación de glucosa a través de su efecto inhibidor sobre el complejo de la piruvato deshidrogenasa lo que, de esta forma, provoca un incremento de la utilización de ácidos grasos24. Tal como se muestra en la figura 5, los valores de ARNm de la PDK4 descendieron un 35% en el corazón de las ratas sometidas a constricción de la aorta comparados con las ratas control. Por el contrario, en el corazón de las ratas tratadas con atorvastatina no se observaron cambios, lo que sugiere que la prevención del incremento en la actividad NF-kB evita cambios en el metabolismo de los ácidos grasos.
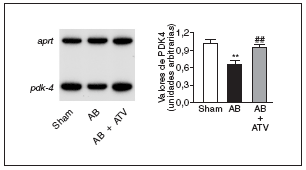
Figura 5. La atorvastatina restablece la expresión de PDK4 en la hipertrofia cardíaca por sobrecarga de presión. Análisis de los valores de ARNm cardíaco en ratas control operadas (sham-operated rats), en ratas con constricción de la aorta (aortic banding [AB]) y en ratas con constricción de la aorta tratadas con atorvastatina (AB + ATV). Se presenta un autorradiograma normalizado respecto al gen control APRT.
Discusión
Aunque existe un gran número de vías de señalización celular implicadas en el desarrollo de la hipertrofia cardíaca25, se conoce muy poco sobre los mecanismos con el potencial suficiente para inhibir o incluso revertir este proceso. En este estudio demostramos que la atorvastatina, igual que otras estatinas15, inhibe la hipertrofia cardíaca inducida por sobrecarga de presión. Además, nuestros resultados indican que las estatinas previenen la activación de NF-kB durante el desarrollo de la hipertrofia cardíaca, lo que puede mejorar la expresión de genes diana PPAR implicados en el metabolismo de los ácidos grasos debido a una reducción de la interacción p65-PPAR.
Los resultados de este estudio demuestran que la inducción de hipertrofia cardíaca, que provoca la activación de NF-kB5, va acompañada de una disminución de la expresión de genes implicados en el metabolismo de los ácidos grasos, como MCAD y PDK4. Los cambios en la expresión de los genes diana de PPAR pueden deberse a una desactivación de PPARα, tal como se ha citado previamente13. Además, hemos demostrado que los valores de expresión de PPARβ/δ, que se expresa en valores comparables a los de PPARα en el corazón14, están reducidos en la hipertrofia cardíaca. Se ha sugerido que PPARα y β/δ comparten funciones similares en el metabolismo cardíaco de los ácidos grasos y que el segundo probablemente compensa la ausencia de PPARα en el músculo esquelético de los ratones PPARα-/-, lo que evita una alteración de la oxidación de los ácidos grasos en estos ratones26. En consecuencia, la reducción de la expresión de ambos tipos de PPAR en el desarrollo de la hipertrofia cardíaca parece ser necesaria para que tenga lugar la disminución de la expresión de los genes implicados en el metabolismo de los ácidos grasos. Cabe destacar que de forma análoga a los resultados obtenidos en este estudio, nuestro grupo ha demostrado previamente que la atorvastatina previene la reducción de la expresión hepática de PPARα en ratas alimentadas con sacarosa27, lo que sugiere que esta estatina puede evitar las alteraciones metabólicas producidas por diferentes estímulos.
Los cambios causados por la hipertrofia cardíaca sobre la expresión de los genes implicados en el metabolismo de los ácidos grasos no se observaron cuando se evitó el aumento de actividad NF-kB con el tratamiento con atorvastatina. Estos resultados parecen implicar a NF-kB en estos cambios. En consonancia con esta idea, un estudio reciente demostró que la inhibición de NF-kB aumentaba la expresión de un gen diana de PPARα, el gen Apo A-I23, lo que confirma la interacción negativa entre NF-kB y PPARα. A continuación, determinamos si además de la citada reducción en la expresión de PPARα durante el desarrollo de la hipertrofia cardíaca13, la interacción física entre PPAR y la subunidad p65 de NF-kB11,12 contribuía a reducir la expresión de genes implicados en el metabolismo cardíaco de los ácidos grasos. Los resultados presentados demuestran que la hipertrofia cardíaca aumenta la asociación proteína-proteína entre p65, por un lado, y PPARα y β/δ, por el otro, lo que indica que este mecanismo puede interferir la capacidad de transactivación de PPAR y contribuir a reducir la expresión de genes implicados en el metabolismo de los ácidos grasos. Por el contrario, prevenir el aumento de la actividad NF-kB mediante el tratamiento con atorvastatina evitó la interacción PPAR-p65 y restableció la expresión de estos genes.
En resumen, los resultados de este estudio demuestran que el tratamiento con atorvastatina inhibe la vía de señalización de NF-kB en la hipertrofia cardíaca inducida por sobrecarga de presión. Además, nuestros resultados señalan que la activación de NF-kB durante el desarrollo de la hipertrofia cardíaca puede contribuir a provocar cambios en el metabolismo de los ácidos grasos a través de un mecanismo que supone un aumento de la interacción proteína-proteína entre los PPAR y la subunidad p65 de NF-kB.