




El aneurisma aórtico abdominal (AAA) es una afección degenerativa y multifactorial caracterizada por una dilatación progresiva de la aorta y activación crónica de inflamación, actividad proteolítica y estrés oxidativo en la pared vascular. La respuesta inmune dependiente de anticuerpos IgG frente a antígenos expuestos en el vaso dañado está implicada en la formación y progresión del AAA, aunque los mecanismos no son del todo conocidos. En este trabajo analizamos la funcionalidad de los receptores Fc de IgG (FcγR), en particular los expresados por el monocito/macrófago, en el desarrollo del AAA experimental.
MétodosEn el modelo de AAA inducido por perfusión aórtica de elastasa se examinaron, mediante histología y PCR cuantitativa, las aortas abdominales de ratones de fenotipo salvaje y de ratones deficientes en FcγR, sin y con transferencia adoptiva de macrófagos. In vitro, macrófagos murinos se transfectaron con ARN de interferencia de FcγRIV/CD16.2 o se trataron con un inhibidor de la cinasa Syk antes de la estimulación con inmunocomplejos de IgG.
ResultadosLa transferencia adoptiva de macrófagos a ratones deficientes en FcγR incrementó su susceptibilidad al desarrollo de AAA. En los ratones que recibieron macrófagos con FcγR funcionales se observó un mayor incremento del diámetro aórtico y del contenido de macrófagos y linfocitos B, así como un aumento en la expresión de la quimiocina CCL2, las citocinas TNF-α e IL-17, la metaloproteinasa MMP2, la enzima prooxidante NADPH oxidasa-2 y las isoformas FcγRIII/CD16 y FcγRIV/CD16.2. In vitro, tanto el silenciamiento génico de FcγRIV/CD16.2 como la inhibición de Syk en macrófagos redujeron la producción de citocinas y anión superóxido inducida por inmunocomplejos.
ConclusionesLa activación de los FcγR en macrófagos contribuye al desarrollo de AAA a través de la inducción de mediadores de inflamación, proteólisis y estrés oxidativo. La modulación de FcγR o sus moléculas efectoras podría ser una potencial diana para el tratamiento del AAA.
Abdominal aortic aneurysm (AAA) is a multifactorial, degenerative disease characterized by progressive aortic dilation and chronic activation of inflammation, proteolytic activity, and oxidative stress in the aortic wall. The immune response triggered by antibodies against antigens present in the vascular wall participates in the formation and progression of AAA through mechanisms not completely understood. This work analyses the function of specific IgG receptors (FcγR), especially those expressed by monocytes/macrophages, in the development of experimental AAA.
MethodsIn the elastase-induced AAA model, the abdominal aortas from wildtype and FcγR deficient mice with/without macrophage adoptive transfer were analysed by histology and quantitative PCR. In vitro, mouse macrophages were transfected with RNA interference of FcγRIV/CD16.2 or treated with Syk kinase inhibitor before stimulation with IgG immune complexes.
ResultsMacrophage adoptive transfer in FcγR deficient mice increased the susceptibility to AAA development. Mice receiving macrophages with functional FcγR exhibited higher aortic diameter increase, higher content of macrophages and B lymphocytes, and upregulated expression of chemokine CCL2, cytokines (TNF-α and IL-17), metalloproteinase MMP2, prooxidant enzyme NADPH oxidase-2, and the isoforms FcγRIII/CD16 and FcγRIV/CD16.2. In vitro, both FcγRIV/CD16.2 gene silencing and Syk inhibition reduced cytokines and reactive oxygen species production induced by immune complexes in macrophages.
ConclusionsActivation of macrophage FcγR contributes to AAA development by inducing mediators of inflammation, proteolysis, and oxidative stress. Modulation of FcγR or effector molecules may represent a potential target for AAA treatment.
El aneurisma de aorta abdominal (AAA) es un proceso degenerativo de la pared vascular en el que se produce la dilatación progresiva de la aorta, generalmente en su porción infrarrenal, considerándose patológica cuando el diámetro del vaso es mayor o igual a 3cm. Los AAA son mayoritariamente asintomáticos y se diagnostican en muchos casos de forma casual mediante pruebas de imagen. Tabaquismo, edad avanzada, sexo masculino, hipertensión e hipercolesterolemia son los principales factores de riesgo del AAA, que presenta una prevalencia del 1-2% en hombres mayores de 65 años. La principal complicación de esta dolencia es la rotura aórtica, que causa hemorragia aguda y una elevada tasa de mortalidad. Actualmente no existe un tratamiento farmacológico capaz de prevenir o limitar la progresión del AAA. En los casos sintomáticos, o cuando el diámetro del aneurisma supera los 5-5,5cm, se realiza una cirugía de reparación abierta o endovascular para mitigar el riesgo de rotura1,2.
La fisiopatología del AAA es compleja, multifactorial y no del todo conocida. Las características patológicas incluyen la degradación progresiva de las fibras elásticas, el infiltrado inflamatorio, la disfunción de las células de músculo liso vascular (CMLV) y el aumento del estrés oxidativo3. En la actualidad se considera el AAA un proceso inflamatorio crónico con participación del sistema inmune innato y adaptativo4. Se sabe que durante el daño vascular se produce el infiltrado gradual de neutrófilos, macrófagos, linfocitos T, mastocitos y células NK, cuya activación libera mediadores inflamatorios y especies reactivas de oxígeno (ROS) y promueve la actividad de metaloproteinasas (MMP) y la apoptosis de CMLV, lo que favorece el debilitamiento de la pared vascular y la rotura del AAA5. En las lesiones de AAA también se han detectado células B productoras de anticuerpos y se han identificado diferentes anticuerpos, predominantemente del isotipo IgG, que son capaces de reconocer proteínas circulantes o componentes de la pared vascular y activar la respuesta inflamatoria en el tejido aneurismático, aunque las moléculas efectoras de esta respuesta autoinmune no se conocen del todo6,7.
Las IgG y los inmunocomplejos (IC) formados por unión antígeno-IgG son capaces de interaccionar con las células inmunes y tisulares a través de receptores específicos para la región constante Fc de IgG (FcγR), activando una amplia gama de respuestas biológicas como fagocitosis, citotoxicidad dependiente de anticuerpos y liberación de mediadores inflamatorios. Desde el punto de vista funcional, estos receptores se clasifican en activadores (en humano, FcγRI/CD64, FcγRIIA,C/CD32A,C y FcγRIIIA,B/CD16A,B; en ratón, FcγRI/CD64, FcγRIII/CD16 y FcγRIV/CD16.2) y el inhibidor FcγRIIB/CD32B (en ratón y humano), que difieren en especificidad, afinidad, distribución celular y vías se señalización. Varias isoformas activadoras se asocian intracelularmente con la cadena-γ, el polipéptido que contiene el motivo de activación ITAM implicado en la transducción de la señal mediante la activación de cinasas Src y Syk8. Estudios en pacientes y modelos experimentales han demostrado que el balance entre isoformas activadoras/inhibidoras de los FcγR y sus ligandos regula diversos procesos implicados en el desarrollo de enfermedades cardiovasculares, entre ellos función endotelial, trombosis, hipertensión, aterosclerosis e inflamación9–11. En el contexto del AAA, recientemente nuestro grupo ha descrito que la deficiencia en la cadena-γ común a varios FcγR activadores limita el desarrollo de lesiones en ratones12. En el presente trabajo analizamos, mediante estudios in vivo de transferencia adoptiva y experimentos in vitro, la posible contribución de los FcγR de macrófagos a la respuesta inflamatoria, la actividad proteolítica y el estrés oxidativo durante la formación del AAA experimental.
Materiales y métodosModelo experimental de AAAEl cuidado y mantenimiento de los animales, así como los procedimientos llevados a cabo en este estudio, se realizaron bajo el principio de las 3R (sustitución, refinamiento o reducción), de acuerdo con la normativa europea (Directiva 2010/63/UE), y fueron aprobados por el Comité Ético de Bienestar Animal de la institución y la comunidad autónoma (PROEX 116/16 y 217/19). Se emplearon ratones deficientes en la cadena-γ (GKO) y controles de fenotipo salvaje (WT), ambos de la cepa C57BL/6J (Jackson Lab). Los ratones se alojaron en jaulas ventiladas (2-4 ratones por jaula) con material de cama habitual y enriquecimiento ambiental en una sala convencional con temperatura controlada (20-22°C), ciclo de luz/oscuridad de 12h y acceso libre a agua y comida estándar. La inducción de AAA se realizó en ratones macho de 10-12 semanas de edad mediante perfusión intraaórtica de elastasa pancreática porcina tipo i (actividad específica 7U/mg; E1250; Sigma-Aldrich)12. Brevemente, los ratones se anestesiaron por inhalación de 2% isoflurano y se realizó una laparotomía horizontal. Utilizando un estereomicroscopio quirúrgico, se separó la aorta abdominal de la vena renal izquierda hasta la bifurcación y se ligó temporalmente entre las arterias renal e ilíaca. Se realizó una aortotomía, se introdujo un tubo de polietileno PE-26 y se perfundió la aorta con elastasa durante 5min a 100mmHg. Luego se reparó la aortotomía, se eliminó la ligadura y se confirmó visualmente la restauración del flujo sanguíneo. Las incisiones se cerraron y los ratones se alojaron en condiciones estándar. Para la transferencia adoptiva de macrófagos, se recolectaron células de médula ósea de fémures de ratones donantes WT y se diferenciaron a macrófagos por incubación en medio DMEM suplementado con 100U/mL de penicilina, 100μg/mL de estreptomicina, 2mM de L-glutamina, 10% de suero bovino fetal (FBS) y 10% de medio condicionado de células L-929. Después de 7 días, los macrófagos se despegaron de las placas de cultivo y se inyectaron 2×106 células por vía intravenosa en ratones receptores GKO, comenzando el día antes de la perfusión de elastasa y cada 4 días durante los siguientes 14 días. El día 14 después de la cirugía, los ratones se anestesiaron (100mg/kg de ketamina y 15mg/kg de xilazina), se perfundieron con salino a través del ventrículo izquierdo, se realizó la eutanasia y se extrajo la aorta. La región infrarrenal de la aorta abdominal se incluyó en parafina para estudios de histología (n=5 ratones por grupo) o se procesó para análisis de expresión génica (n=4 ratones por grupo).
Análisis histológicoSecciones transversales seriadas de 4μm de la aorta abdominal (a lo largo de aproximadamente 200μm) se desparafinaron y tiñeron de forma alternada con tricrómico de Masson. Para cada ratón se midió el diámetro aórtico en el punto máximo de la lesión, definiendo el AAA como un aumento≥100% del diámetro. Para el análisis inmunohistoquímico, las muestras se desparafinaron y rehidrataron, se realizó la recuperación antigénica con tampón citrato y se inactivó la peroxidasa endógena con 3% H2O2:metanol (1:1). Después de bloquear las uniones inespecíficas (PBS con 4% de albúmina y 8% de suero del huésped del anticuerpo secundario), las muestras se incubaron durante 16h a 4°C con anticuerpo primario frente a macrófagos (CD68, Abcam Cat# ab53444, RRID:AB_869007) y linfocitos B (CD45R, BD Biosciences Cat# 550286, RRID:AB_393581). Tras un lavado, las secciones se incubaron con anticuerpo secundario biotinilado, seguido del complejo avidina-biotina-peroxidasa y su cromógeno (diaminobenzidina o aminoetilcarbazol), y finalmente se contrastaron con hematoxilina. La tinción positiva se cuantificó de forma ciega en al menos 2 secciones por ratón utilizando el software Image-Pro Plus y se expresó en porcentaje de área positiva con respecto al área total de la lesión.
Cultivos celulares, inhibición y silenciamientoLa línea celular de macrófagos murinos RAW 264.7 (TIB-71, ATCC) se mantuvo en medio DMEM suplementado con antibióticos, L-glutamina y 10% de FBS. Las células se mantuvieron en medio sin FBS durante 16h, se pretrataron durante 1h con el inhibidor de Syk (Bay 613606, 500nM) y se estimularon durante 6 y 24h con 150μg/mL de agregados solubles de IgG. Los agregados de IgG se obtuvieron por calentamiento de IgG murina (10mg/mL, 30min a 63°C) y posterior centrifugación. Para el silenciamiento génico, las células se transfectaron en medio Opti-MEM con 20nmol de ARN pequeño de interferencia (ARNip) específico para FcγRIV/CD16.2 (siCD16.2; sc-42759, Santa Cruz Biotech) o con un ARNip inespecífico como control negativo (siScr, 4390843, Thermo Fisher) utilizando el reactivo Lipofectamine RNAiMAX (13778075, Thermo Fisher). El silenciamiento se comprobó mediante Western blot con anticuerpo anti-FcγRIV/CD16.2 (Santa Cruz Biotech) y α-tubulina (Sigma-Aldrich) como control de carga.
Expresión de ARNmEl ARN total de aorta y células se extrajo con el reactivo TRI (Molecular Research Center). A partir de 1μg de ARN se obtuvo el ADNc mediante reacción de transcripción inversa utilizando el kit High Capacity cDNA Archive (Applied Biosystems) en un termociclador convencional (PTC-100, MJ Research Inc.) bajo las siguientes condiciones: 10min a 25°C, 2h a 37°C y 5min a 85°C. Los niveles de expresión de los genes diana se determinaron por duplicado mediante PCR cuantitativa en tiempo real (qPCR) empleando el sistema de detección secuencial TaqMan ABI 7700 con TaqADN polimerasa activada por calor y cebadores comerciales murinos (Ccl2, Mm00441242_m1; Tnfα, Mm00443258_m1; Il17, Mm00439618_m1; Mmp2, Mm00439498_m1; Nox2, Mm01287743_m1; Nox4, Mm00479246_m1; Fcγr1/Cd64, Mm00438874_m1; Fcγr3/Cd16, Mm00438883_m1; Fcγr4/Cd16.2, Mm00519988_m1; Applied Biosystems), realizando un paso inicial de 2min a 50°C y 10min a 95°C y 40 ciclos posteriores de 15s a 95°C y 1min a 60°C. Los resultados de la amplificación del gen de interés se normalizaron con el control interno 18S ARNr amplificado en paralelo. La cantidad de ARNm se estimó mediante el método de cuantificación 2-ΔCT.
ELISALos niveles de fosforilación de Syk (P-Syk) se determinaron mediante ELISA celular. Brevemente, las células se sembraron en placas de 96 pocillos (105cél/pocillo) y se estimularon a diferentes tiempos con IC en presencia o ausencia del inhibidor de Syk. Después se fijaron con 4% de formaldehído, se bloquearon (PBS con 2% de albúmina y 5% de FBS) y se incubaron con anticuerpos anti-P-Syk (Y323, Y317; 44-234G, Invitrogen) o anti-Syk (2712, Cell Signaling) durante 16h a 4°C, seguido de anticuerpo secundario conjugado con peroxidasa y el sustrato 3,3’,5,5’-tetrametilbenzidina. La absorbancia se midió en un lector de placas (λ=450nm y corrección a 570nm). Los valores de P-Syk se corrigieron por los de Syk total (analizado en pocillos paralelos) y el contenido de células por pocillo (tinción con azul de metileno). La concentración de las proteínas CCL2 y TNF-α secretadas al medio de cultivo se cuantificó empleando kits de ELISA (MJE00, R&D Systems; 88-7324-22, Thermo Fisher Scientific).
Medida de estrés oxidativoLa producción de anión superóxido dependiente de NADPH oxidasa (NOX) se determinó mediante ensayo de quimioluminiscencia, como se indicó anteriormente13. Las células estimuladas se resuspendieron en tampón fosfato (50mM de K2HPO4/KH2PO4, 0,01mM de EDTA, 0,32M de sacarosa y 0,1% de cóctel de inhibidores de proteasas) y se transfirieron a tubos Röhren. La reacción quimioluminiscente se inició por adición secuencial de 5μM de lucigenina y 100μM de NADPH. La emisión de luz por oxidación de la lucigenina se cuantificó cada 10s durante 10min en un luminómetro Sirius (Berthold), expresando los valores en unidades de quimioluminiscencia por miligramo de proteína celular.
Análisis estadísticoLos resultados se presentan como datos individuales y media±desviación estándar (DE) del total de animales o experimentos individuales. El análisis estadístico se realizó con el programa GraphPad v. 8 (GraphPad Software Inc.) empleando la prueba ANOVA de una vía con comparaciones múltiples de Tukey. Se estableció significación estadística para valores de p<0,05.
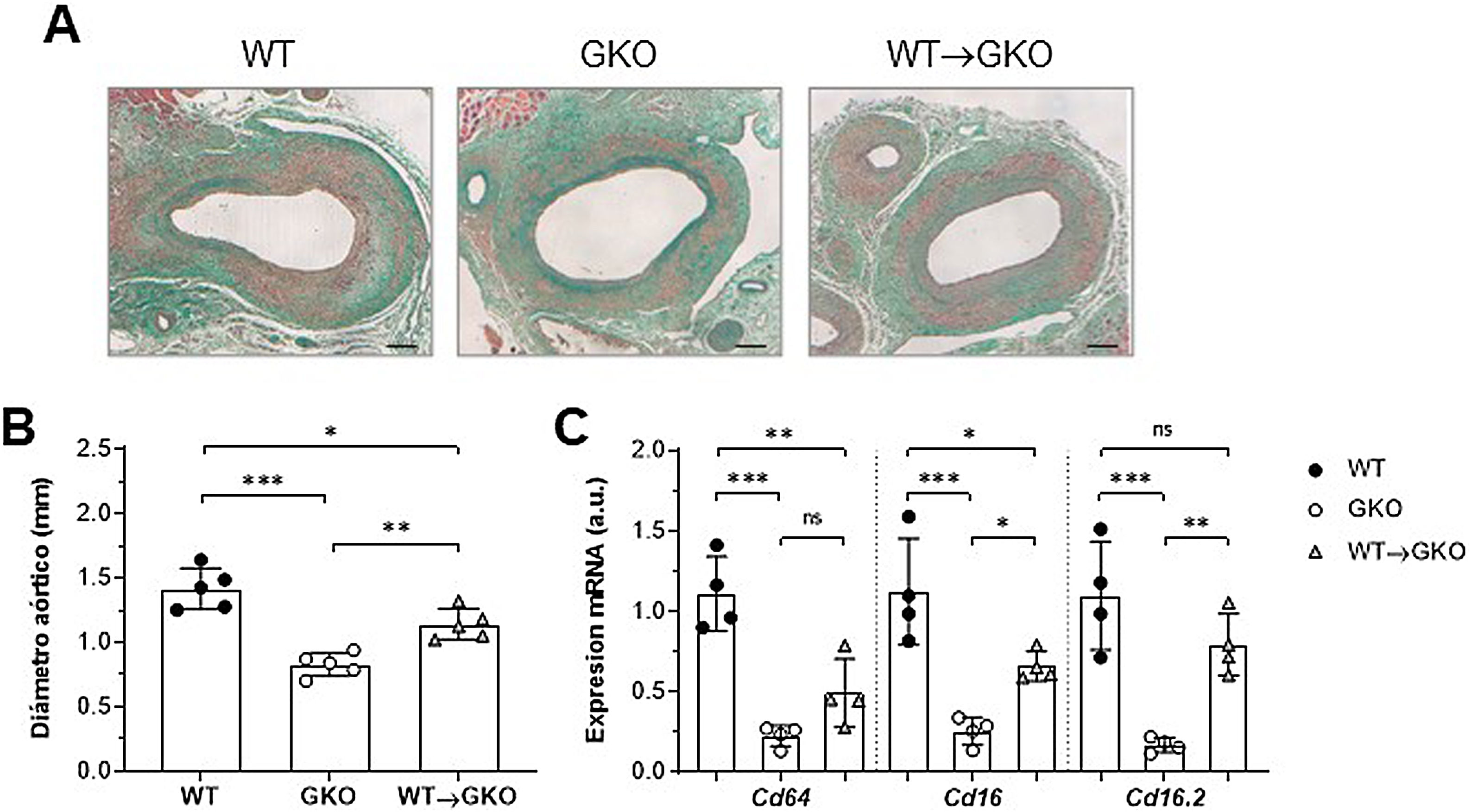
ResultadosLa activación de FcγR en macrófagos agrava el desarrollo de AAA en ratonesNuestro estudio previo demostró que la respuesta inmunoinflamatoria producida en la lesión aneurismática promueve la expresión de isoformas FcγR en la pared vascular del AAA humano y que la disfunción de FcγR protege frente al desarrollo de AAA experimental12. Para profundizar en el papel específico de los FcγR del monocito/macrófago en la formación del AAA, realizamos un modelo combinado de transferencia adoptiva e inducción de AAA con elastasa. Para ello, células de médula ósea aisladas de ratones WT se diferenciaron a macrófagos y se transfirieron a ratones GKO (deficientes en la cadena-γ asociada a FcγRI/CD64, FcγRIII/CD16 y FcγRIV/CD16.2), a los que se indujo AAA mediante perfusión con elastasa. A los 14 días se realizó el estudio histológico en cortes consecutivos de la aorta abdominal de ratones (fig. 1A). En comparación con el grupo WT, los ratones GKO mostraron menor incidencia de AAA (60 vs. 100%) y menor diámetro aórtico (% vs. WT: 42±9; fig. 1B). Por el contrario, en los ratones GKO que recibieron macrófagos WT (grupo WT→GKO) se observó una incidencia de AAA del 100% y un aumento significativo del diámetro aórtico con respecto a ratones GKO sin transferencia adoptiva (% vs. GKO: 173±28%; fig. 1B). Mediante análisis de qPCR observamos que la transferencia de macrófagos WT restauró parcialmente la expresión de FcγR activadores (fig. 1C), destacando la isoforma FcgRIV/CD16.2, que alcanzó niveles próximos al grupo WT.
 de la tinción de tricrómico de Masson (A) y medida del diámetro aórtico (B) en secciones de aorta abdominal de ratones de fenotipo salvaje (WT), deficientes en cadena-γ (GKO) y con transferencia adoptiva de macrófagos WT (WT→GKO) a los 14 días de la perfusión aórtica de elastasa. (C) Análisis mediante qPCR de la expresión de ARNm de isoformas activadoras FcγR. Datos normalizados por 18S ARNr y expresados en unidades arbitrarias (a.u.). Valores individuales y media±DE de 4-5 ratones por grupo.")
La presencia de FcγR activadores en macrófagos agrava el desarrollo de AAA inducido por elastasa en ratones. Imágenes representativas (barras de escala, 100μm) de la tinción de tricrómico de Masson (A) y medida del diámetro aórtico (B) en secciones de aorta abdominal de ratones de fenotipo salvaje (WT), deficientes en cadena-γ (GKO) y con transferencia adoptiva de macrófagos WT (WT→GKO) a los 14 días de la perfusión aórtica de elastasa. (C) Análisis mediante qPCR de la expresión de ARNm de isoformas activadoras FcγR. Datos normalizados por 18S ARNr y expresados en unidades arbitrarias (a.u.). Valores individuales y media±DE de 4-5 ratones por grupo.
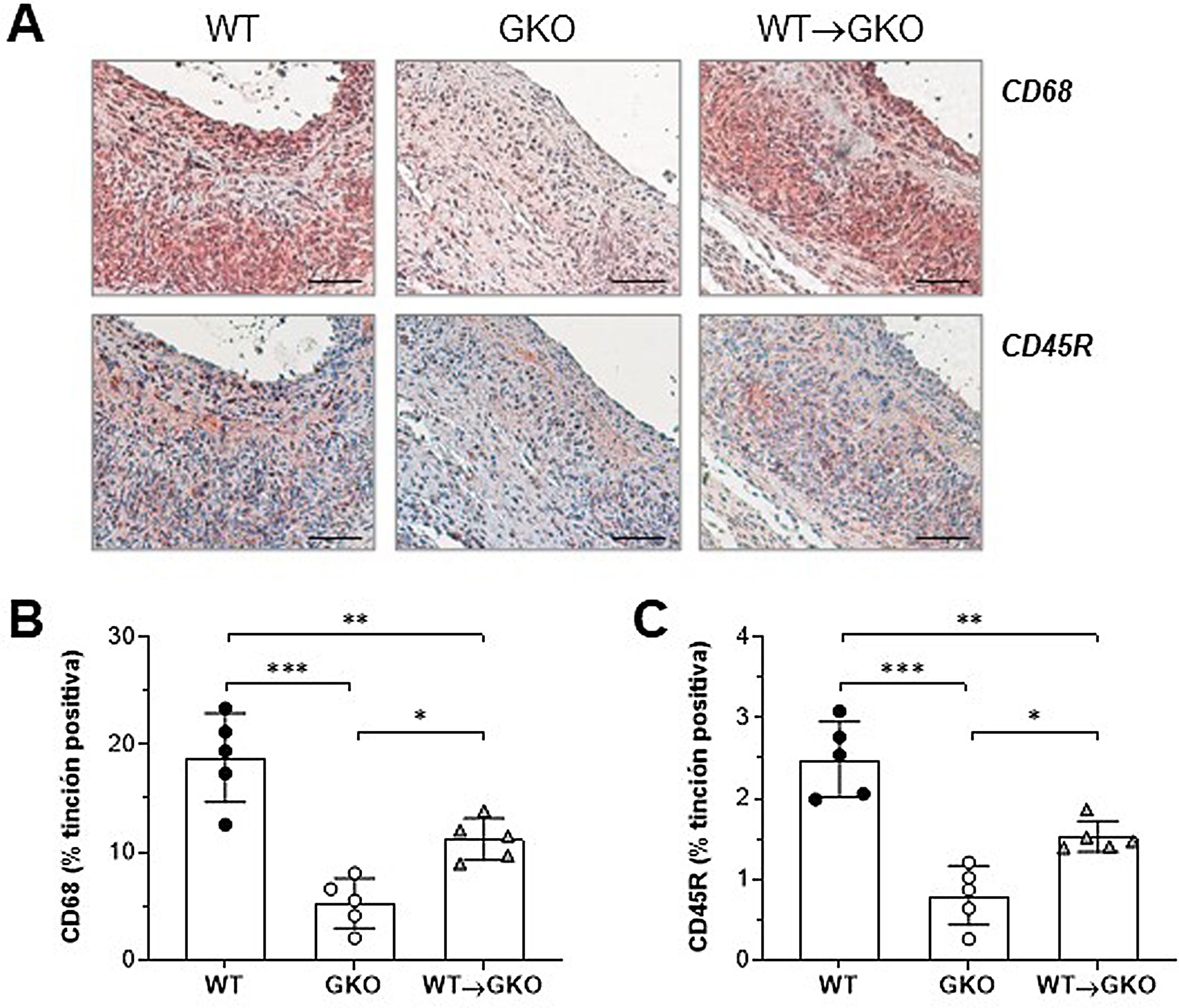
El análisis inmunohistoquímico de macrófagos (marcador CD68) y linfocitos B (marcador CD45R) mostró un aumento significativo del contenido de ambos tipos de células inflamatorias en las lesiones del grupo WT→GKO con respecto al grupo GKO (% vs. GKO: 212±37 y 191±21, respectivamente), aunque estos valores fueron inferiores a los del grupo control WT (fig. 2). Estos resultados evidencian una mayor respuesta inflamatoria en la lesión aneurismal asociada a la presencia de macrófagos con FcγR funcionales.
 de la tinción inmunohistoquímica (A) y cuantificación del área de tinción positiva de macrófagos CD68+ (B) y linfocitos B CD45B+ (C) en lesiones AAA de ratones de los grupos WT, GKO y WT→GKO. Valores individuales y media±DE de 5 ratones por grupo.")
La transferencia adoptiva de macrófagos con FcγR activadores aumenta el infiltrado inflamatorio en las lesiones AAA. Imágenes representativas (barras de escala, 50μm) de la tinción inmunohistoquímica (A) y cuantificación del área de tinción positiva de macrófagos CD68+ (B) y linfocitos B CD45B+ (C) en lesiones AAA de ratones de los grupos WT, GKO y WT→GKO. Valores individuales y media±DE de 5 ratones por grupo.
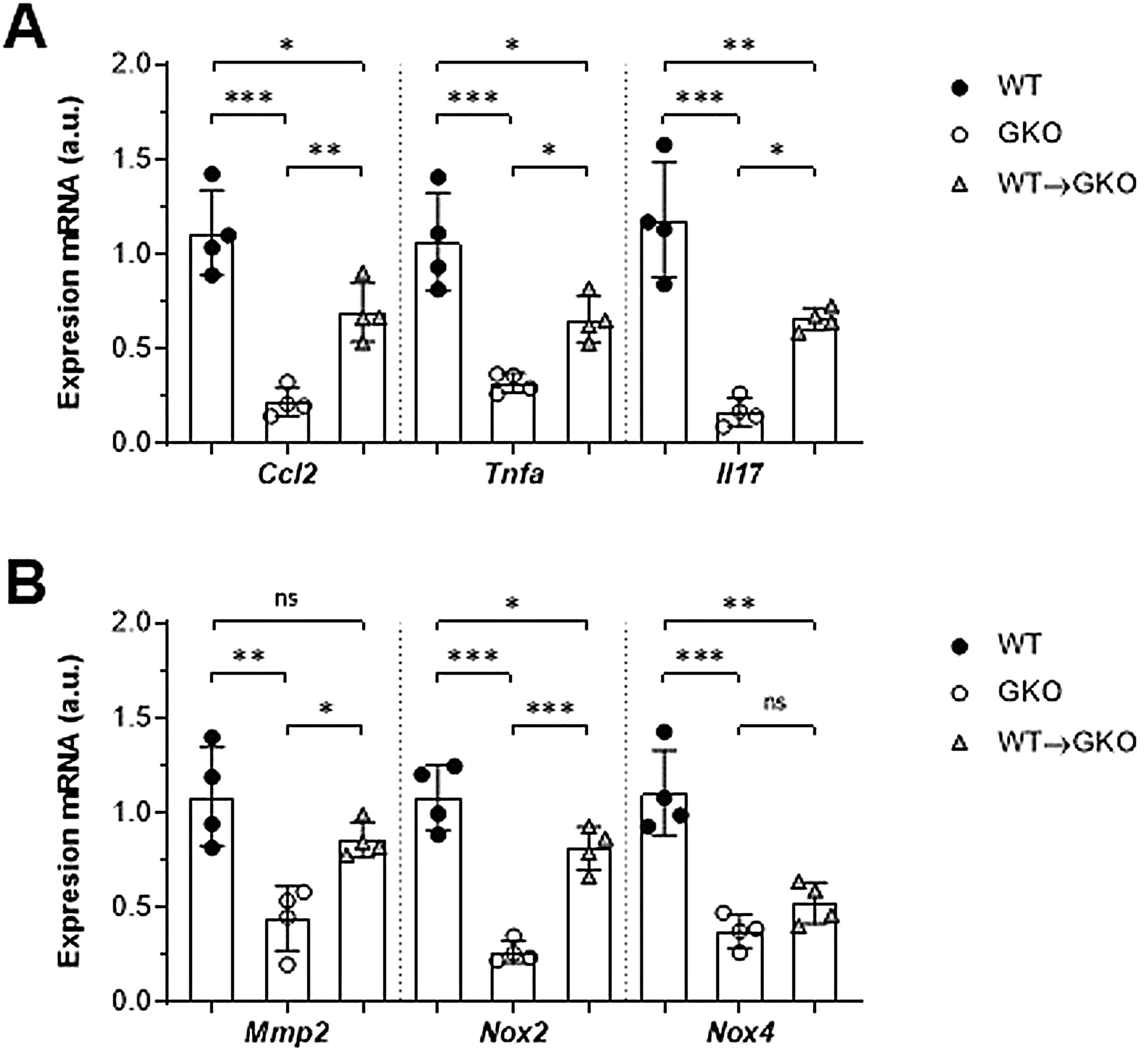
La expresión de genes implicados en la patogenia del AAA se analizó mediante qPCR. Observamos un importante descenso en la expresión génica de la quimiocina CCL2 y las citocinas TNF-α e IL-17 en la aorta abdominal de ratones GKO en comparación con los WT (% vs. WT: 20±7, 30±5 y 14±6, respectivamente; fig. 3A). Por el contrario, la transferencia de macrófagos WT incrementó de forma significativa estos niveles de expresión (% vs. GKO: 314±70, 204±37 y 395±35, respectivamente; fig. 3A). En el grupo WT→GKO también observamos un aumento en los niveles de expresión de la MMP2 y de la enzima prooxidante NOX2 (% vs. GKO: 194±21 y 305±43, respectivamente), mientras que la expresión de la isoforma NOX4 no se modificó de forma significativa (fig. 3B).
 y enzimas proteolíticas y prooxidantes (B) en muestras murinas de los grupos WT, GKO y WT→GKO. Datos normalizados por 18S ARNr y expresados en unidades arbitrarias (u.a.). Valores individuales y media±DE de 4 ratones por grupo.")
Impacto de la transferencia de macrófagos con FcγR en la expresión de genes en aorta abdominal. Análisis mediante qPCR de quimiocinas y citocinas (A) y enzimas proteolíticas y prooxidantes (B) en muestras murinas de los grupos WT, GKO y WT→GKO. Datos normalizados por 18S ARNr y expresados en unidades arbitrarias (u.a.). Valores individuales y media±DE de 4 ratones por grupo.
Estos resultados indican que la transferencia adoptiva de macrófagos con FcγR funcionales en los ratones deficientes en FcγR agrava el proceso inflamatorio y oxidativo e incrementa la susceptibilidad al desarrollo de AAA.
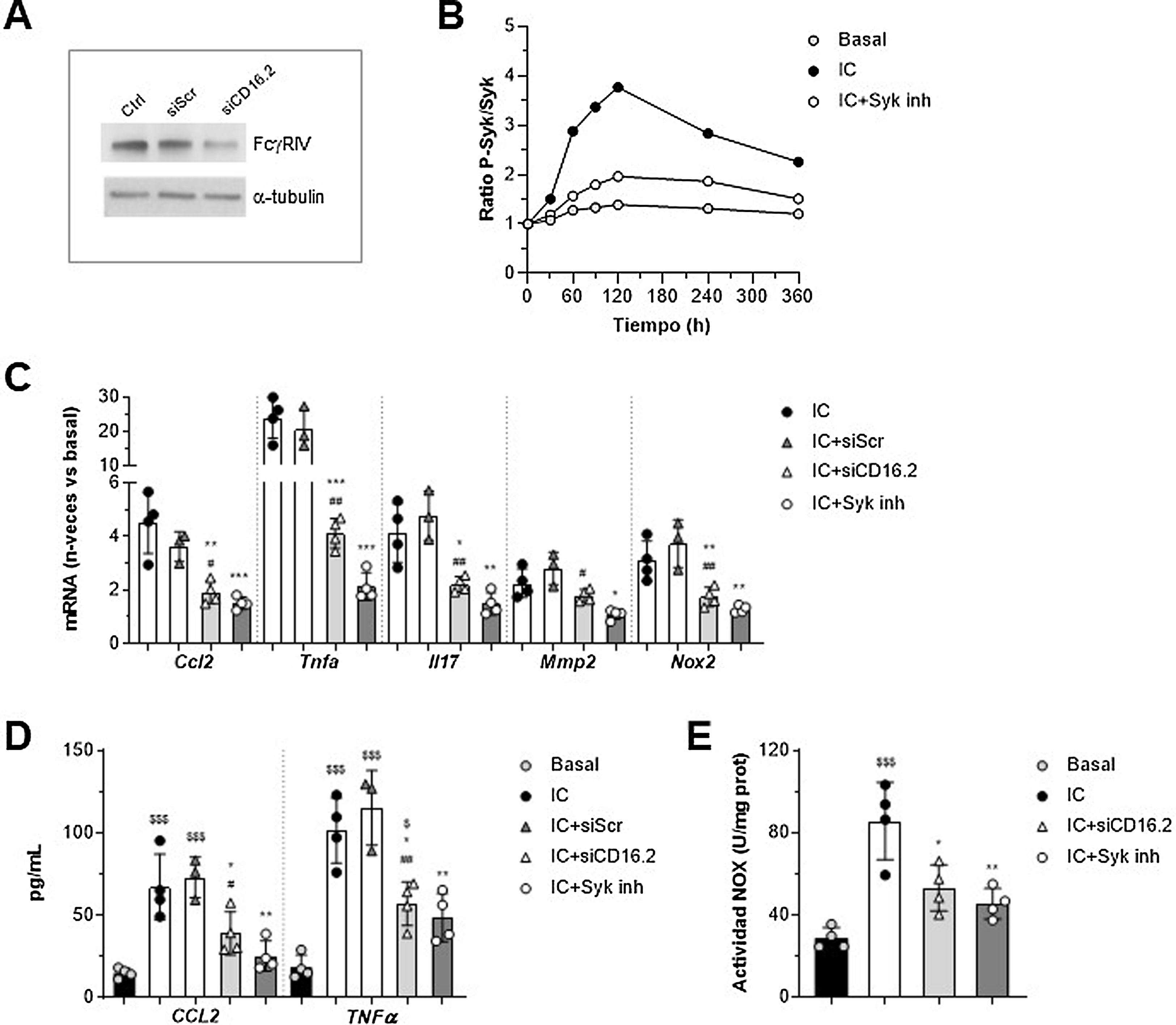
Implicación de FcγRIV/CD16.2 y Syk en la respuesta de los macrófagos in vitroPara profundizar en las funciones dependientes de FcγR en el contexto del AAA, establecimos un sistema in vitro en macrófagos murinos estimulados con agregados solubles de IgG como símil de los IC, ya que presentan propiedades parecidas a estos y excluyen la contribución del antígeno. Por una parte, puesto que la transferencia de macrófagos restauró mayoritariamente los niveles de FcγRIV/CD16.2 en aorta, realizamos experimentos de silenciamiento génico de esta isoforma para determinar su contribución a las acciones de los IC. Por otra parte, evaluamos la implicación de la tirosina cinasa Syk, un mediador clave en la vía de señalización de FcγR activadores. La supresión de FcγR/CD16.2 en células tratadas con ANAip específico para FcgRIV (siCD16.2) en comparación con la condición inespecífica o scramble (siScr) se confirmó por Western blot (fig. 4A). Mediante ELISA celular comprobamos que el tratamiento con el inhibidor selectivo Bay 613606 previno la fosforilación de Syk inducida por IC a lo largo del tiempo (fig. 4B).
 y su control negativo (siScr) o se pretrataron con el inhibidor Bay 613606 y posteriormente se estimularon con IC (agregados de IgG, 150μg/mL). (A) Inmunoblot representativo de FcgRIV/CD16.2 y α-tubulina en células sin transfectar (Ctrl) y transfectadas. (B) ELISA celular de Syk (ratio fosforilada/total) en células tratadas con el inhibidor de Syk. Experimento representativo realizado en cuadruplicado. (C) Análisis de la expresión de ARNm mediante qPCR. Valores normalizados con 18S ARNr y expresados en incremento vs. condiciones basales (n=3-4 experimentos). (D) Análisis mediante ELISA de la secreción de proteínas CCL2 y TNF-α al sobrenadante celular (n=3-4 experimentos). (E) Medida de la producción de ROS dependiente de NOX mediante ensayo de lucigenina, expresada en unidades de quimioluminiscencia por concentración de proteína (n=4 experimentos). *p<0,05, **p<0,01 y ***p<0,001 vs. IC; #p<0,05 y ##p<0,01 vs. siScr; $p<0,05 y $$$p<0,001 vs. basal.")
Implicación de FcγRIV/CD16.2 y Syk en las respuestas mediadas por inmunocomplejos in vitro. Macrófagos RAW 264.7 se transfectaron con ARNip específico de FcγRIV/CD16.2 (siCD16.2) y su control negativo (siScr) o se pretrataron con el inhibidor Bay 613606 y posteriormente se estimularon con IC (agregados de IgG, 150μg/mL). (A) Inmunoblot representativo de FcgRIV/CD16.2 y α-tubulina en células sin transfectar (Ctrl) y transfectadas. (B) ELISA celular de Syk (ratio fosforilada/total) en células tratadas con el inhibidor de Syk. Experimento representativo realizado en cuadruplicado. (C) Análisis de la expresión de ARNm mediante qPCR. Valores normalizados con 18S ARNr y expresados en incremento vs. condiciones basales (n=3-4 experimentos). (D) Análisis mediante ELISA de la secreción de proteínas CCL2 y TNF-α al sobrenadante celular (n=3-4 experimentos). (E) Medida de la producción de ROS dependiente de NOX mediante ensayo de lucigenina, expresada en unidades de quimioluminiscencia por concentración de proteína (n=4 experimentos). *p<0,05, **p<0,01 y ***p<0,001 vs. IC; #p<0,05 y ##p<0,01 vs. siScr; $p<0,05 y $$$p<0,001 vs. basal.
El análisis de qPCR mostró una reducción significativa de la expresión de ARNm de CCL2, TNF-α, IL-17, MMP2 y NOX2 en macrófagos transfectados con siCD16.2 y estimulados con IC en comparación con las células sin transfectar o transfectadas con siScr (fig. 4C). De forma similar, la inhibición de Syk previno de forma importante la expresión de genes inducida por los IC (fig. 4C). Ambos tratamientos también redujeron la secreción de proteínas CCL2 y TNF-α al medio extracelular de los macrófagos (fig. 4D). Por último, mediante ensayo de lucigenina observamos que la incubación de macrófagos con IC promueve la generación de anión superóxido dependiente de NOX; dicho efecto se encontró atenuado en células silenciadas con siCD16.2 o tratadas con el inhibidor de Syk (fig. 4E).
Estos resultados in vitro confirman el papel directo de la isoforma FcγRIV/CD16.2 y de la tirosina cinasa Syk en la respuesta inflamatoria y oxidativa de los macrófagos a la estimulación con IC.
DiscusiónLa inflamación, el estrés oxidativo y la degradación de la matriz extracelular, junto con la activación del sistema inmune, son procesos clave en la patogenia del AAA. Varios estudios evidencian que la activación de la respuesta autoinmune frente a autoantígenos depositados o expuestos en la pared vascular está implicada en la formación y progresión del AAA, aunque los mecanismos moleculares subyacentes no están del todo aclarados4. El presente trabajo demuestra que los receptores de IgG FcγR, en particular las isoformas activadoras presentes en el macrófago, son mediadores de la respuesta inmunoinflamatoria durante el desarrollo del AAA experimental. Nuestros hallazgos demuestran que: a) la deficiencia funcional de FcγR activadores en ratones GKO limita el desarrollo de AAA; b) la reconstitución con macrófagos que expresan FcγR agrava el proceso inflamatorio y la formación de AAA, y c) las moléculas FcγRIV/CD16.2 y Syk son clave en la respuesta inflamatoria y oxidativa mediada por IC en los macrófagos.
La presencia de linfocitos B e Ig en lesiones de AAA evidencia un papel de la inmunidad humoral mediada por anticuerpos como impulsor de la formación y progresión del AAA14. Diversos estudios han detectado niveles elevados de IgG, IgA, IgE e IgM en el suero y/o la pared vascular de pacientes con AAA y modelos experimentales15. En pacientes con AAA se ha encontrado asociación entre los niveles (en plasma y tejido) de IgG anti-HDL y el diámetro aórtico16, y los niveles circulantes de anticuerpos antifosfolípidos correlacionan con marcadores de inflamación y predicen de forma independiente la progresión del AAA17. Además, la fracción de IgG extraída de lesiones de AAA presenta reactividad frente a diversos componentes del tejido conectivo y es capaz de activar el sistema del complemento, participando así en el desarrollo de AAA7,18. En ratones, la deficiencia en células B reduce la presencia de Ig en las lesiones y protege frente el desarrollo de AAA19. En este trabajo mostramos un menor infiltrado de células B en las lesiones AAA de ratones deficientes GKO y un aumento en los ratones reconstituidos con macrófagos que expresan FcγR, lo que evidencia la implicación de estos receptores en el reclutamiento y/o activación de linfocitos B productores de anticuerpos en la enfermedad.
Estudios previos en ratones deficientes en una o múltiples isoformas de FcγR han demostrado el papel central de las interacciones IgG-FcγR en diversas enfermedades autoinmunes e inflamatorias8. En enfermedades cardiovasculares, se ha propuesto que la generación de autoanticuerpos dirigidos frente a epítopos propios (circulantes o de la pared vascular) puede contribuir a la inflamación vascular y el remodelado tisular a través de varios mecanismos, entre los que destacan la opsonización y el aclaramiento por fagocitos a través de FcγR, así como la formación y/o depósito de IC capaces de activar el sistema del complemento y la respuesta de leucocitos y de células tisulares15. Numerosos estudios en modelos murinos de aterosclerosis han demostrado que el bloqueo de isoformas FcγR activadoras reduce el tamaño de las placas de ateroma10,11,20, mientras que la ausencia del receptor inhibidor agrava el proceso21. Sin embargo, los hallazgos en el contexto del AAA son más limitados. En pacientes con AAA, los niveles del receptor FcγRIII/CD16 en monocitos circulantes se asocian con los de biomarcadores inflamatorios y un mayor tamaño de las lesiones22, y en aorta se ha encontrado el FcγRIIB/CD32B localizado en zonas de infiltrado inflamatorio23. En nuestro estudio previo, la comparación de lesiones AAA humanas con aortas sanas demostró expresión aumentada de todas las isoformas FcγR. También detectamos la colocalización de las isoformas activadoras FcγRI/CD64 y FcgRIIA/CD16A con los depósitos de IgG en zonas de la pared vascular ricas en macrófagos y CMLV, evidenciando que tanto las células infiltrantes como las residentes del vaso pueden participar en la respuesta inmune durante la formación del AAA12. En modelos experimentales de AAA, el análisis de secuenciación identificó la regulación positiva de genes asociados a la vía de «fagocitosis mediada por FcγR en el monocito/macrófago»24 y la deleción génica de receptores Fc para IgE25 y para IgG12 limita la formación de AAA en modelos de infusión de angiotensina ii o de elastasa. Los hallazgos del presente trabajo indican que la transferencia adoptiva de macrófagos revierte el fenotipo resistente de los ratones deficientes en FcγR frente al desarrollo de AAA, como demuestra el incremento en el tamaño de las lesiones AAA, el mayor contenido de células inflamatorias y la expresión elevada de isoformas FcγR, citocinas, metaloproteinasas y enzimas prooxidantes. Estos resultados resaltan la contribución de los FcγR del monocito/macrófago a la respuesta inflamatoria y oxidativa y al remodelado vascular patológico durante el AAA.
La activación de monocitos/macrófagos participa en todas las fases de formación de AAA, desde el desarrollo inicial hasta la aparición de disección o rotura aórtica26. Los principales factores involucrados en la acumulación de macrófagos en la pared del AAA incluyen quimiocinas y citocinas producidas en respuesta a una lesión tisular, productos de degradación de la matriz extracelular y producción de ROS, entre otros5. Estos factores del microambiente local también influyen en la respuesta y el perfil de expresión de los macrófagos, promoviendo su polarización hacia un amplio espectro de fenotipos con funciones tanto patogénicas como reparadoras del tejido. Durante las primeras etapas de formación de lesiones AAA se produce un incremento gradual de macrófagos de fenotipo proinflamatorio M1 que contribuye al estrés oxidativo, la inflamación y el remodelado vascular, mientras que el fenotipo antiinflamatorio M2 se ha asociado con fases avanzadas, posiblemente como un mecanismo compensador para prevenir la expansión y/o rotura del AAA26,27. En monocitos humanos, diversas citocinas proinflamatorias y antiinflamatorias, así como el estado redox, modulan el balance entre FcγR activadores e inhibidores y, por tanto, la magnitud de sus respuestas28,29. Por otra parte, se ha reportado que la activación de FcγRIIA/CD32A y FcγRIII/CD16 promueve la polarización M130, mientras que FcγRIIB/CD32B se asocia con un fenotipo M2b productor de citocinas antiinflamatorias31. En este trabajo, la transferencia adoptiva de macrófagos con FcγR favorece la acumulación de células inflamatorias en las lesiones e incrementa la expresión de la quimiocina CCL2, de las citocinas inflamatorias TNF-α e IL-17 y de la enzima proteolítica MMP2, todos mediadores típicos de la respuesta inflamatoria M1 y el remodelado vascular. También observamos un marcado aumento de NOX2, la isoforma fagocítica mayoritariamente implicada en la generación de ROS por los macrófagos. Sin embargo, no se alteró la expresión de NOX4, isoforma asociada a células no fagocíticas como células endoteliales y CMLV32. Teniendo esto en cuenta, aunque nuestro estudio destaca la importancia de los FcγR del monocito/macrófago en la formación del AAA, no podemos descartar que otros tipos celulares contribuyan al proceso patológico. De hecho, tanto las células endoteliales como las CMLV expresan varias isoformas de FcγR, cuya estimulación promueve una variedad de respuestas celulares como producción de citocinas y ROS, apoptosis, proliferación y quimiotaxis10,12,33,34.
Desde el punto de vista del mecanismo, este estudio implica a la isoforma FcγRIV/CD16.2 y la cinasa Syk en las respuestas mediadas por los IC en el contexto del AAA, lo que evidencia que ambas moléculas podrían ser potenciales dianas de actuación en la enfermedad. El receptor murino FcγRIV es ortólogo del humano FcγRIIIA/CD16A, interacciona con afinidad intermedia con las subclases más potentes de IgG (IgG2a e IgG2b) y participa en la citotoxicidad celular dependiente de anticuerpo y fagocitosis. Los ratones deficientes en FcγRIV/CD16.2 están protegidos frente al desarrollo de diversas enfermedades inmunes, mientras que el bloqueo de su actividad inhibe la liberación de mediadores inflamatorios en neutrófilos y macrófagos35. Nuestros resultados demuestran por vez primera que la transferencia de macrófagos in vivo recupera los niveles de expresión de la isoforma FcγRIV/CD16.2 en las lesiones AAA y que su silenciamiento génico in vitro previene la expresión de genes proinflamatorios y prooxidantes, así como la liberación de ROS inducida por los IC.
Por otra parte, se sabe que la agregación de FcγR activadores por IC en la superficie celular de los leucocitos activa la familia de tirosina cinasa Src, que fosforila los motivos ITAM en la cadena-γ asociada al receptor, promoviendo la fosforilación y activación de Syk y la subsiguiente activación de múltiples vías de señalización8. Syk también participa en la señalización de receptores de antígenos de linfocitos B y T y de receptores de lectina de tipo C, que también contienen motivos ITAM y se encuentran activados en diversas enfermedades inmunes e inflamatorias36. En muestras de AAA de pacientes y modelos experimentales se han encontrado incrementos en la fosforilación de Syk en la pared vascular37. In vitro, Syk regula la diferenciación de macrófagos a fenotipo M1, la proliferación y migración celular y la secreción de mediadores inflamatorios38, y su inhibición suprime las respuestas de linfocitos y CMLV y limita la progresión del AAA12,37. Confirmando estas publicaciones previas, nuestro estudio con el inhibidor selectivo de Syk evidencia que la activación del eje FcγR-Syk participa en la respuesta inflamatoria y oxidativa del macrófago a los IC en el contexto del AAA.
En conclusión, este trabajo indica que la interacción entre los IC y los FcγR activadores presentes en el macrófago participa en la patogenia del AAA a través de la inducción de mediadores de inflamación, proteólisis y estrés oxidativo. Estos hallazgos apoyan el uso de terapias inmunomoduladoras dirigidas a los FcγR o sus moléculas efectoras para inhibir el daño vascular mediado por la inmunidad humoral en el AAA.
FinanciaciónEstudio financiado por la Sociedad Española de Arteriosclerosis (BIB05/18), el Ministerio de Ciencia e Innovación (RTI2018-098788-B-I00 y PID2021-127741OB-I00) y La Caixa Foundation (HR17-00247), y premiado como mejor comunicación póster en el xxxiv congreso de la SEA.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
Los autores agradecen a Ana Melgar y Patricia Quesada (IIS-FJD/UAM) su apoyo técnico en los estudios histológicos.