





El envejecimiento es el principal factor de riesgo de enfermedad cardiovascular (ECV) y su prevalencia está aumentando progresivamente debido en gran parte al incremento de la esperanza de vida a nivel mundial. En este contexto, es fundamental establecer cuáles son los mecanismos por los que el envejecimiento promueve el desarrollo de ECV, con el objetivo de reducir su incidencia. La aterosclerosis y la insuficiencia cardiaca contribuyen de manera significativa a la morbimortalidad por ECV asociada a la edad. El síndrome de progeria de Hutchinson-Gilford (HGPS) se caracteriza por un envejecimiento prematuro que cursa también con ECV acelerada. Se trata de un trastorno genético raro causado por la expresión de progerina, una forma mutada de la prelamina A. La progerina induce aterosclerosis masiva y alteraciones electrofisiológicas en el corazón, promueve el envejecimiento y finalmente la muerte prematura a una edad media de 14,6 años, principalmente por infarto de miocardio o ictus cerebral. En esta revisión se discuten las principales alteraciones estructurales y funcionales que afectan al sistema vascular durante el envejecimiento fisiológico y prematuro, así como los mecanismos que subyacen a la aterosclerosis y al envejecimiento exagerados inducidos por la prelamina A y la progerina. Dado que ambas proteínas se expresan en individuos sin HGPS y muchas de las características del envejecimiento normal se presentan en la progeria, la investigación en el ámbito del HGPS podría contribuir a la identificación de nuevos mecanismos implicados en el envejecimiento cardiovascular fisiológico.
Aging is the main risk factor for cardiovascular disease (CVD). The increased prevalence of CVD is partly due to the global increase in life expectancy. In this context, it is essential to identify the mechanisms by which aging induces CVD, with the ultimate aim of reducing its incidence. Both atherosclerosis and heart failure significantly contribute to age-associated CVD morbidity and mortality. Hutchinson-Gilford progeria syndrome (HGPS) is a rare genetic disorder caused by the synthesis of progerin, which is noted for accelerated aging and CVD. This mutant form of prelamin A induces generalised atherosclerosis, vascular calcification, and cardiac electrophysiological abnormalities, leading to premature aging and death, mainly due to myocardial infarction and stroke. This review discusses the main vascular structural and functional abnormalities during physiological and premature aging, as well as the mechanisms involved in the exacerbated CVD and accelerated aging induced by the accumulation of progerin and prelamin A. Both proteins are expressed in non-HGPS individuals, and physiological aging shares many features of progeria. Research into HGPS could therefore shed light on novel mechanisms involved in the physiological aging of the cardiovascular system.
Las enfermedades cardiovasculares (ECV) están íntimamente relacionadas con el envejecimiento y son responsables de más de un 30% de la mortalidad mundial1,2. Aunque el envejecimiento está asociado al desarrollo de un amplio abanico de enfermedades, las ECV constituyen la mayor carga para la población anciana, sus cuidadores y los sistemas de salud3. La elevada prevalencia de las ECV se deriva de la mejora en su tratamiento, lo que ha incrementado la esperanza de vida en los países desarrollados y ha contribuido al envejecimiento progresivo de la población. Si bien este hecho supone un increíble éxito desde el punto de vista individual, el cambio demográfico asociado representa un reto para los sistemas de protección social y asistencia sanitaria de todo el mundo. Por ejemplo, en Estados Unidos se calcula que la población de más de 65 años pasará del 12% en 2010 al 22% en el año 20404. En 2050 se prevé que 19 países tengan más de un 10% de su población por encima de los 80 años, afectada en gran parte por ECV u otras enfermedades asociadas al envejecimiento y dependiente del trabajo de terceros5. El coste económico del tratamiento de los pacientes con ECV es enorme y se prevé que se incremente sustancialmente en los próximos años. Por ejemplo, los estados miembros de la Unión Europea gastan colectivamente más de 4 billones de euros diarios en atención sanitaria6. Asimismo, en Estados Unidos está previsto que se triplique el coste médico directo de las ECV entre 2010 y 2030, y que se incrementen un 61% los costes indirectos debido a la pérdida de productividad asociada a estas enfermedades4. Por tanto, existe una necesidad apremiante de identificar los mecanismos por los que el envejecimiento induce el deterioro del sistema cardiovascular, independientemente de otros factores de riesgo, gran parte de los cuales son modificables. Este conocimiento es esencial para poder ofrecer asistencia médica eficaz y sostenible a una población que envejece rápidamente.
Características comunes del envejecimiento fisiológico y el síndrome de progeria de Hutchinson-GilfordSe han identificado 4factores principales como causantes del daño acumulativo responsable del envejecimiento en mamíferos: la inestabilidad genómica, el acortamiento de los telómeros, las alteraciones epigenéticas y la pérdida de la proteostasis7. Estos procesos inducen los llamados mecanismos antagónicos, que comprenden alteraciones en la sensibilidad a los nutrientes, disfunción mitocondrial y senescencia celular. Estos, a su vez, promueven el desarrollo de los llamados mecanismos integrales, que son los principales promotores del envejecimiento, e incluyen el agotamiento de las células madre y la alteración de la comunicación intercelular. Estos mecanismos de envejecimiento se han identificado, o bien mediante análisis comparativos entre individuos o animales jóvenes y aquellos que han envejecido normalmente, o bien como resultado de estudios de intervención en los que se analiza cómo afecta a la esperanza de vida la alteración específica de vías genéticas o procesos bioquímicos durante el envejecimiento fisiológico.
Existen evidencias de que la investigación en el síndrome de progeria de Hutchinson-Gilford (HGPS) (código OMIM 176670) podría contribuir al conocimiento de los mecanismos celulares y moleculares que promueven el envejecimiento normal y las ECV asociadas. Los pacientes con HGPS muestran un envejecimiento prematuro asociado a una aceleración de la aterosclerosis y la calcificación vascular, junto con el desarrollo de alteraciones en la conductividad eléctrica del miocardio. Estas alteraciones suelen conllevar la muerte de los pacientes en los primeros años de la adolescencia (esperanza de vida de 14,6 años), principalmente debido a infarto de miocardio o ictus cerebral. La enfermedad está causada por la expresión de progerina, una variante anómala de la prelamina A que se genera como consecuencia de una mutación de novo en el gen LMNA8,9 (véase más abajo). Se han descrito otros síndromes progeroides asociados a mutaciones que conllevan la pérdida de función en el gen ZMPSTE24 y una acumulación anómala de prelamina A10. Cabe destacar que todos los factores característicos del envejecimiento normal propuestos por López-Otín et al.7 se han descrito en modelos animales de progeria, algunos de los cuales se han detectado también en pacientes con HGPS7,11-14. Asimismo, el envejecimiento normal en individuos sin HGPS se caracteriza por bajos niveles de expresión de prelamina A y progerina en diversos tipos celulares y tejidos11-13, incluyendo células de la adventicia, la media y lesiones ateroscleróticas en arterias coronarias15. Se ha descrito que tanto el estrés oxidativo como el acortamiento de los telómeros, fenómenos que se considera que contribuyen al envejecimiento normal7, promueven la expresión de la prelamina A y la progerina en células normales16,17. En esta revisión se resumen las principales alteraciones estructurales y funcionales del sistema vascular durante el envejecimiento, así como los mecanismos moleculares y celulares implicados, basándonos en nuestra reciente revisión sobre cómo el envejecimiento afecta al sistema cardiovascular en su conjunto18.
Alteraciones vasculares en el envejecimiento fisiológicoAlteraciones del sistema nervioso autónomoEl envejecimiento provoca alteraciones importantes en la regulación que ejerce el sistema nervioso autónomo sobre el sistema cardiovascular. Estas alteraciones incluyen un incremento de la actividad simpática debido al aumento de los niveles de catecolaminas en sangre, así como una disminución de la sensibilidad β-adrenérgica, y una disminución del reflejo barorreceptor en las arterias19-21. Aunque todavía se desconocen las causas concretas de estas alteraciones, algunos autores han propuesto que la disminución de la sensibilidad β-adrenérgica y de la actividad de los barorreceptores arteriales podría desencadenar el incremento de la actividad simpática como mecanismo compensatorio22,23.
HipertensiónLa premisa clásica de que el envejecimiento causa hipertensión sigue siendo en la actualidad motivo de intenso debate22. Resultados procedentes de estudios longitudinales han mostrado que la actividad simpática asociada al envejecimiento incrementa la presión arterial19; sin embargo, son los valores iniciales de tensión arterial en la juventud los que condicionan que este aumento supere o no el umbral para ser considerada hipertensión24. Sun et al. destacan el papel etiológico de la rigidez vascular en la elevación de la presión sanguínea y proponen que la rigidez de las arterias de gran calibre disminuye la actividad de los barorreceptores, que finalmente se compensa con un incremento de la actividad simpática, lo que provoca una subida de la tensión arterial23. La regulación aguda, a corto plazo (beat to beat), de la presión arterial se lleva a cabo mayoritariamente mediante los barorreflejos. Puesto que la edad se asocia con una disminución en la actividad de los barorreflejos, el envejecimiento se ha asociado también a un incremento de la variación en la presión arterial25,26.
Rigidez vascular, disfunción endotelial y aterosclerosisLa rigidez arterial y la disfunción endotelial son 2de las principales alteraciones vasculares relacionadas con la edad1-3.
La disfunción endotelial conlleva alteraciones en el control del tono y la permeabilidad vascular, lo que favorece los procesos proinflamatorios3. El óxido nítrico, uno de los principales mediadores celulares liberado por el endotelio, ve reducida su biodisponibilidad, lo cual aumenta la permeabilidad y la inflamación endotelial, y desencadena un proceso de retroalimentación positiva que agrava el fenómeno a largo plazo27. La disfunción endotelial juega un papel primordial tanto en el inicio como en la progresión de la aterosclerosis y forma parte del proceso degenerativo que afecta a las grandes arterias durante el envejecimiento2,22,28. Así, el envejecimiento también está asociado con un remodelado vascular que implica un aumento de la luz del vaso y un engrosamiento de la túnica íntima y de la túnica media29. El incremento de la relación íntima-media es un síntoma temprano de aterosclerosis22 y es un predictor independiente de futuros eventos cardiovasculares30. El engrosamiento de la íntima se inicia y mantiene debido al reclutamiento de leucocitos sanguíneos, proceso desencadenado por la activación de moléculas de adhesión en las células endoteliales (CE) disfuncionales31. Los leucocitos en la neoíntima inducen una respuesta inmune local compleja que promueve un mayor reclutamiento de leucocitos e induce la migración de las células musculares lisas de la pared vascular (CMLV) desde la túnica media hacia la lesión aterosclerótica en crecimiento. Las CMLV activadas en la neoíntima se transforman de un fenotipo «contráctil» a uno «sintético», caracterizado por desdiferenciación, proliferación y secreción abundante de componentes de la matriz extracelular32,33.
Existe una importante controversia acerca de si la aterosclerosis es el resultado de la acumulación de factores de riesgo con el envejecimiento o, por el contrario, el envejecimiento por sí mismo promueve aterosclerosis independientemente de otros factores. La ausencia de aterosclerosis entre los ancianos de sociedades tribales aisladas34 y su presencia en niños con un alto grado de exposición35 atestigua la importante influencia de la exposición a factores de riesgo y señala que es posible envejecer sin aterosclerosis. Sin embargo, se han detectado signos de aterosclerosis en restos de momias humanas procedentes de sociedades no expuestas a los factores de riesgo modernos36. Este hecho, junto con la incidencia de aterosclerosis en individuos con HGPS y otros síndromes de envejecimiento prematuro, apoyan la hipótesis de que el factor de riesgo aterosclerótico más importante es el envejecimiento en sí mismo.
La rigidez arterial es otra característica patológica propia del envejecimiento que se considera que promueve el inicio y progresión de la hipertensión y aterosclerosis al inducir disfunción de las CE y las CMLV de la capa media37. La rigidez vascular se puede medir de manera no invasiva a partir de la velocidad de onda de pulso. Este parámetro es un indicador de eventos cardiacos muy fiable e independiente de la presión arterial en diversas poblaciones adultas, incluyendo los ancianos38. La velocidad de onda de pulso carotidofemoral aumenta progresivamente a partir de los 50 años de edad y puede alcanzar una incidencia de un 64% en hombres y un 74% en mujeres de más de 70 años39,40. El aumento de la rigidez de las grandes arterias incrementa el esfuerzo cardiaco e induce fibrosis e insuficiencia cardiaca.
Entre los mecanismos que contribuyen al aumento de la rigidez arterial durante el envejecimiento destacan las alteraciones de la matriz extracelular y el aumento asociado de fibrosis e inflamación23. La modificación de la estructura y composición de la matriz extracelular de la pared vascular en respuesta a la edad se debe al aumento en la deposición y maduración del colágeno, la acumulación de productos finales de glicación avanzada y la rotura de las fibras elásticas20,37,41.
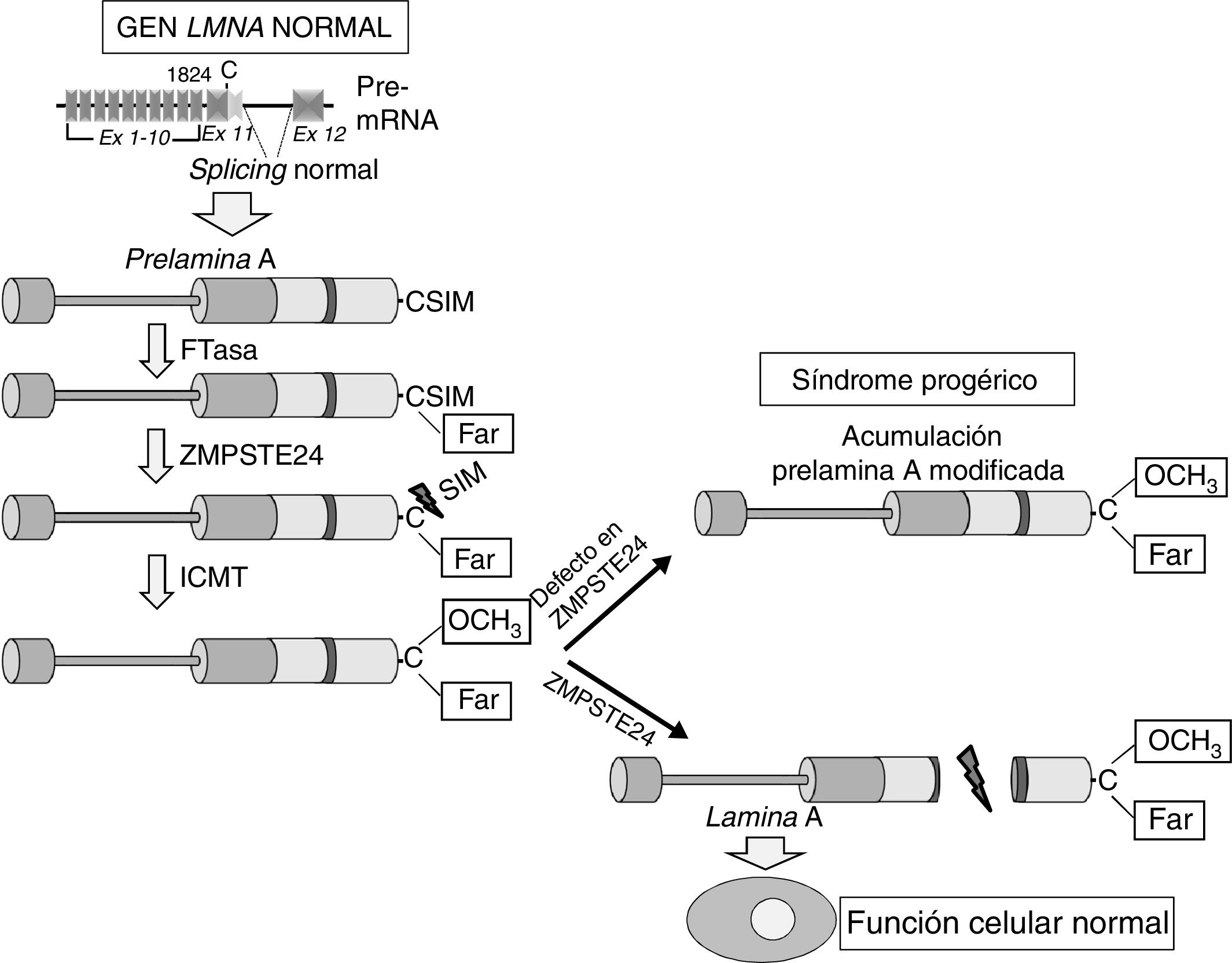
Enfermedad cardiovascular en pacientes con síndrome de progeria de Hutchinson-GilfordLa HGPS es una enfermedad genética humana ultrarrara con una prevalencia estimada de una por cada 20 millones de personas (www.progeriaresearch.org). La enfermedad se caracteriza por un envejecimiento acelerado causado por una mutación de novo en el gen LMNA8,9. En células normales, el splicing alternativo del tránscrito primario del ARNm del gen LMNA da lugar a 2variantes principales de lamina de tipo A (lamina A y lamina C), además de variantes menos abundantes (la lamina AΔ10 y la lamina C2, específica de células germinales)42-44. La proteína precursora de la lamina A, denominada prelamina A, sufre diversas modificaciones postraduccionales para dar lugar a la proteína madura (fig. 1). En primer lugar, una farnesiltransferasa farnesila el residuo de cisteína en el dominio C-terminal cisteína-serina-isoleucina-metionina. A continuación, se eliminan los 3 aminoácidos en posición C-terminal, lo que permite la metilación del nuevo extremo C-terminal catalizado por la isoprenilcisteína carboxil metiltransferasa (ICMT). Finalmente, la metaloproteinasa dependiente de zinc ZMPSTE24/FACE-1 elimina los 15 residuos C-terminales junto con el grupo farnesil y carboximetil. La lamina A madura se incorpora entonces a la lámina nuclear, una red de proteínas estructurales que se asocia íntimamente a la membrana nuclear interna. Además de proporcionar resistencia mecánica al núcleo, las laminas de tipo A regulan múltiples funciones celulares, incluyendo la replicación y reparación del ADN, la organización de la cromatina, la transducción de señales y la transcripción génica45.

Síntesis y procesamiento de la prelamina A en células normales. El splicing normal entre los exones 11 y 12 del gen LMNA permite la síntesis de la prelamina A, la cual sufre una serie de modificaciones postraduccionales que culminan en la producción de la lamina A madura. El procesamiento proteolítico de la prelamina A catalizado por la proteasa ZMPSTE24 elimina su extremo C-terminal farnesilado y carboximetilado. Las mutaciones que inactivan ZMPSTE24 producen la acumulación de prelamina A permanentemente farnesilada y carboximetilada y aceleran el envejecimiento celular. Ex: exón; Far: residuo farnesilado; FTasa: farnesil transferasa; ICMT: isoprenilcisteína carboxil metiltransferasa.
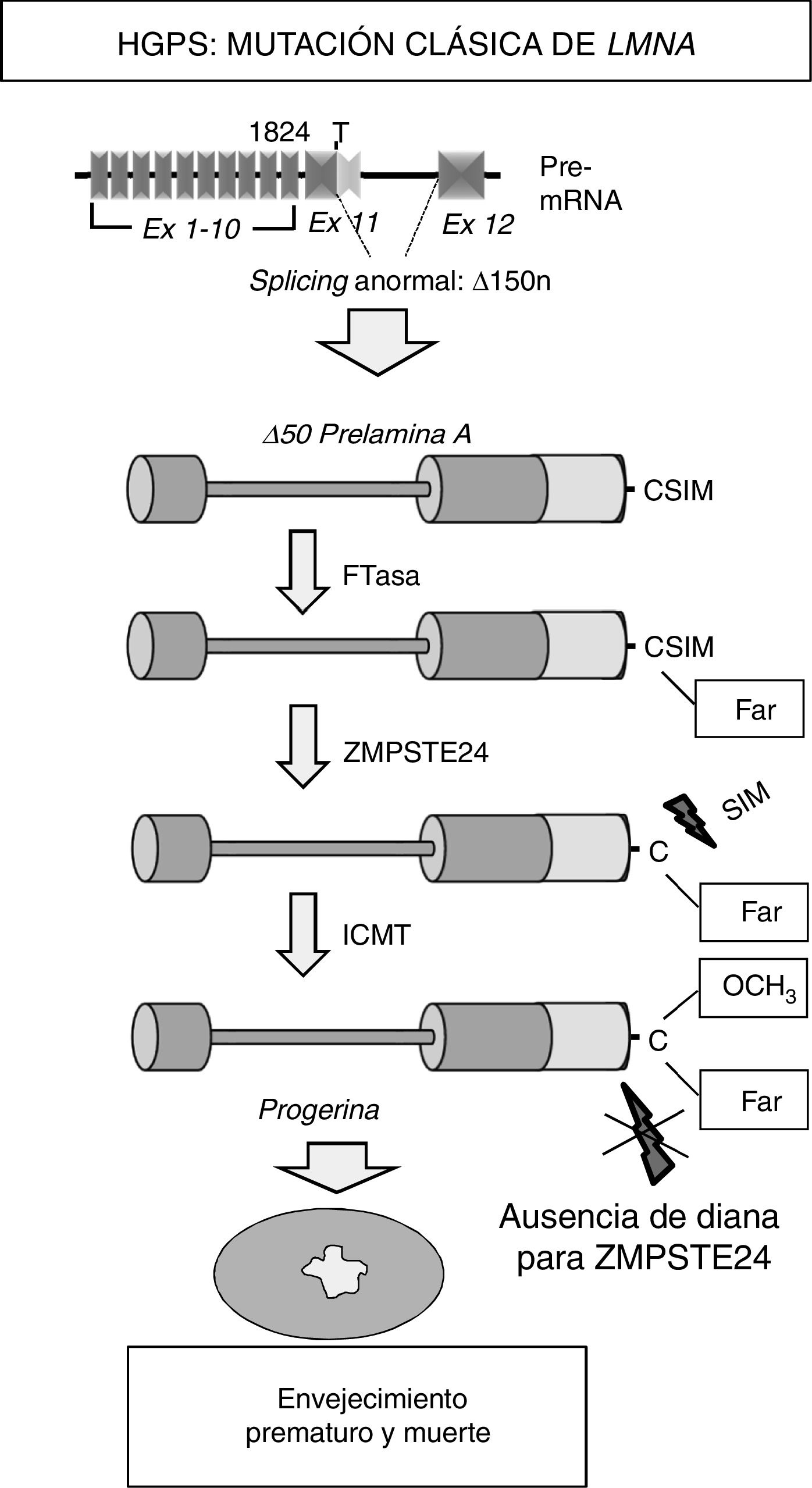
La mayoría de los pacientes HGPS presentan en heterocigosis la mutación puntual de novo c.1824C>T (p.G608G) en el gen LMNA8,9. Aunque esta mutación responsable de la progeria «clásica» es sinónima, crea un sitio de splicing aberrante en el exón 11 que elimina 150 nucleótidos y da lugar a la síntesis de una variante truncada de la prelamina A denominada progerina (Δ50 prelamina A) (fig. 2). La falta de los 50 aminoácidos en el extremo C-terminal de la progerina impide la escisión de los 15 aminoácidos terminales, por lo que las células de los pacientes acumulan progerina permanentemente farnesilada y carboximetilada. A su vez, mutaciones que inactivan el gen ZMPSTE24/FACE-1 también provocan la acumulación de prelamina A farnesilada y carboximetilada y están asociadas con el desarrollo de síndromes progeroides en humanos10.
 que provoca un splicing anómalo y la síntesis de la progerina. La ausencia de la región de 50 aminoácidos que incluye el sitio de reconocimiento para ZMPSTE24 impide la eliminación del extremo C-terminal de la progerina, la cual permanece permanentemente farnesilada y causa múltiples alteraciones celulares que conducen al envejecimiento prematuro y la muerte. Ex: exón; Far: residuo farnesilado; FTasa: farnesil transferasa; ICMT: isoprenilcisteína carboxil metiltransferasa.")
Síntesis y procesamiento de la prelamina A en pacientes con HGPS. La progeria clásica se asocia con una mutación heterocigota puntual de novo en el gen LMNA (c.1824C>T) que provoca un splicing anómalo y la síntesis de la progerina. La ausencia de la región de 50 aminoácidos que incluye el sitio de reconocimiento para ZMPSTE24 impide la eliminación del extremo C-terminal de la progerina, la cual permanece permanentemente farnesilada y causa múltiples alteraciones celulares que conducen al envejecimiento prematuro y la muerte.
Ex: exón; Far: residuo farnesilado; FTasa: farnesil transferasa; ICMT: isoprenilcisteína carboxil metiltransferasa.
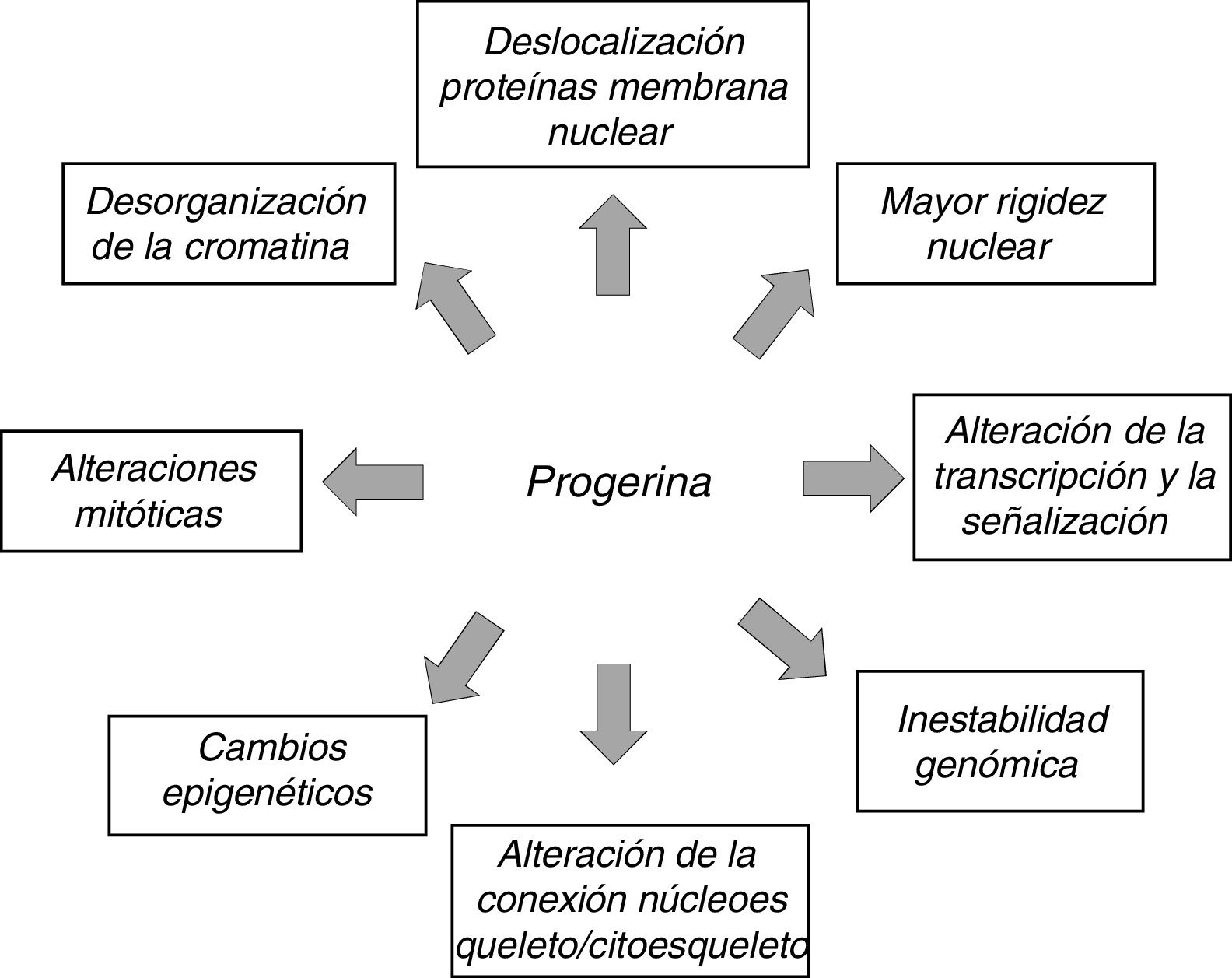
La expresión anómala de prelamina A y progerina causa múltiples alteraciones funcionales y estructurales que afectan a la transducción de señales, la transcripción génica y la organización de la cromatina y, en última instancia, ralentizan la proliferación celular, provocan senescencia y muerte celular y aceleran el envejecimiento del organismo (fig. 3)12,45. Cabe destacar que muchos de los procesos que intervienen en la HGPS están también implicados en el envejecimiento normal7.

Los pacientes con HGPS tienen una apariencia normal al nacer, pero empiezan a desarrollar síntomas durante los primeros 12-18 meses de vida. La enfermedad se caracteriza por un retraso en el crecimiento, dentición anormal, alopecia, lipodistrofia, anomalías de la piel, contracturas articulares, osteoporosis y osteolisis que progresivamente conllevan dificultades para caminar y desarrollar otras actividades motoras. Sin embargo, los problemas médicos más graves en los pacientes con HGPS son la aterosclerosis y las anomalías en la conductividad eléctrica del miocardio, que causan la muerte prematura a una edad media de 14,6 años, mayoritariamente por infarto de miocardio o ictus46-50.
Muchas de las alteraciones cardiovasculares en pacientes con HGPS también se manifiestan en el envejecimiento fisiológico (tabla 1). Sin embargo, a diferencia de los ancianos, los pacientes con HGPS muestran elevación del recuento de plaquetas y del tiempo de protrombina49. Las causas y consecuencias fisiológicas de estas alteraciones se desconocen por el momento, por lo que hacen falta estudios más específicos que puedan determinar su posible relevancia. Además, los pacientes de HGPS suelen carecer de factores de riesgo cardiovascular tradicionales o están solo parcialmente afectados por ellos. Por ejemplo, los pacientes con HGPS tienen niveles de colesterol plasmático, colesterol LDL y HDL, triglicéridos y proteína C reactiva similares a los de los niños sanos48,49,51. Además, aproximadamente el 30% de los pacientes con HGPS presentan solo incrementos ligeros en la presión sistólica y diastólica comparados con niños sanos de la misma edad48,49,52. Por tanto, el estudio de la ECV asociada a HGPS ofrece una oportunidad única para identificar los mecanismos que causan el daño cardiovascular asociado a la edad en ausencia de otros factores de riesgo, o de otras enfermedades crónicas relacionadas con el envejecimiento, que podrían afectar de forma secundaria a la salud cardiovascular.
Cambios estructurales y funcionales del sistema vascular durante el envejecimiento fisiológico y prematuro
| Parámetro | Envejecimiento fisiológico | Envejecimiento prematuro |
|---|---|---|
| Aumento de la actividad simpática | + | + |
| Menor sensibilidad barorrefleja | + | + |
| Engrosamiento íntima-media | + | + |
| Rigidez vascular | + | + |
| Fibrosis vascular | + | + |
| Disfunción endotelial | + | - |
| Aterosclerosis | + | + |
| Calcificación vascular | + | + |
| Hipertensión | + | + |
| Recuento plaquetario elevado | - | + |
| Aumento del tiempo de protrombina | - | + |
+: presencia, -: ausencia.
Al igual que en el envejecimiento normal, las técnicas de imagen no invasiva en pacientes con HGPS tan solo detectan placas carotídeas evidentes en estadios avanzados48,49,52. Sin embargo, la estenosis arterial afecta a pacientes con HGPS de todas las edades y se ha propuesto que podría ser un indicador temprano de formación de placa48. Las placas ateroscleróticas de pacientes con HGPS van acompañadas por otras alteraciones vasculares clásicas del envejecimiento normal, tales como inflamación, pérdida de CMLV y fibrosis en la media, y erosión y rotura de placas15. Sin embargo, una característica de los vasos de pacientes con HGPS poco frecuente en vasos de sujetos control es la presencia de un importante engrosamiento de la adventicia y fibrosis15,53. Otra característica del envejecimiento fisiológico que también se manifiesta en pacientes con HGPS es la calcificación vascular, la cual se asocia con aumento de la morbimortalidad por ECV en la población general54. La calcificación afecta a la aorta y a las válvulas aórtica y mitral de algunos pacientes con HGPS y puede provocar regurgitación aórtica o mitral15,49,55-58. Los estudios de neuroimagen en una cohorte de 25 pacientes detectaron la presencia de ictus tempranos y clínicamente silentes, lo que señala que se trata de una característica prevalente en la HGPS59. Sin embargo, el ictus en pacientes con HGPS puede también provocar secuelas neurológicas60.
La HGPS también se caracteriza por la rigidez vascular, una alteración asociada con el envejecimiento fisiológico que predice la incidencia de eventos cardiovasculares futuros de forma independiente a otros factores38. El análisis de una cohorte de 21 pacientes con HGPS determinó que la rigidez vascular es un fenómeno temprano y generalizado en esta enfermedad: se detectaron valores de velocidad de onda de pulso comparables a los que se presentan habitualmente en adultos de más de 60 años48. Si bien el grosor íntima-media en la arteria carótida de pacientes con HGPS es normal, su ecodensidad es superior a lo normal, en especial en la adventicia, en consonancia con la fibrosis vascular detectada en autopsias15,48,49,61. Los pacientes con HGPS presentan alteraciones del índice tobillo-brazo, un indicador de enfermedad arterial periférica y disfunción vascular48,56; sin embargo, la evaluación no invasiva de la función endotelial mediante la medición de la vasodilatación mediada por flujo parece indicar que los pacientes no presentan disfunción endotelial49. Estos datos ponen de manifiesto la necesidad de realizar estudios adicionales en los que se analicen con detalle los mecanismos que desencadenan la disfunción vascular inducida por la progerina.
En resumen, las características principales de la ECV en pacientes con HGPS son la rigidez y el remodelado vascular, con una importante fibrosis de la media y la adventicia, pérdida de CMLV, aterosclerosis acelerada y muerte prematura por infarto de miocardio o ictus. La muerte prematura en la HGPS también puede ir asociada a arritmias derivadas de las alteraciones en la conductividad eléctrica cardiaca, un aspecto que queda fuera del ámbito de esta revisión12,18. Se requieren nuevos estudios para definir con precisión los mecanismos por los cuales la expresión de la progerina acelera las ECV y para elucidar la contribución relativa de las alteraciones vasculares y cardiacas a la muerte prematura en la HGPS.
Mecanismos y tratamiento del síndrome de progeria de Hutchinson-GilfordLas laminas A/C juegan un papel fundamental en un amplio rango de funciones celulares, incluyendo el mantenimiento de la estabilidad mecánica del núcleo, la transducción de señales, la transcripción génica, la organización de la cromatina, la reparación del ADN, la progresión del ciclo celular y la diferenciación y migración celulares45. La acumulación de progerina y prelamina A, por tanto, acelera el envejecimiento debido a diversos mecanismos que afectan a múltiples vías y no son mutuamente excluyentes. Estos mecanismos activados por prelamina A y progerina podrían diferir entre distintos tejidos en función de las diferencias en la expresión de lamina A (y, por tanto, de prelamina A y progerina), la cual se relaciona con la rigidez tisular62.
Estudios en humanos y en ratones señalan que la gravedad de la HGPS está determinada tanto por la cantidad total de progerina como por la proporción de progerina farnesilada frente a la lamina A madura. De hecho, las distintas mutaciones puntuales de la LMNA humana están asociadas con diferencias importantes en los niveles de progerina y en la gravedad de la enfermedad, que va desde la progeria neonatal (niveles de progerina elevados) hasta la progeria tardía (niveles bajos de progerina)63-65. En concordancia, en ratones LmnaG609G/G609G, portadores de una mutación en el gen endógeno de la Lmna equivalente a la mutación c.1824C>T (p.G608G) que causa la HGPS en humanos, se observó una mejora en el fenotipo de envejecimiento y un aumento de la supervivencia cuando se redujo la expresión de progerina con morfolinos antisentido dirigidos contra el splicing anómalo entre los exones 11 y 1266. Análogamente, el tratamiento con oligonucleótidos antisentido diseñados para promover el splicing alternativo de lamina A hacia lamina C también redujo la producción de progerina, la fibrosis de la adventicia y la pérdida de CMLV en ratones LmnaG609G/G609G, aunque no se ha descrito ningún efecto de esta estrategia sobre la longevidad67.
A diferencia de la lamina A madura, la progerina permanece farnesilada de forma permanente (fig. 1). La hipótesis de que la farnesilación persistente es la principal causa de la progeria se basa en estudios en ratones y en humanos que han demostrado una asociación positiva entre los síntomas progeroides y la acumulación de prelamina A farnesilada derivada de la deficiencia en ZMPSTE2468-71. Asimismo, un paciente que presentaba tanto una mutación homocigota de pérdida de función en ZMPSTE24 como una mutación heterocigota del gen LMNA, que daba lugar a una elongación del extremo C-terminal de la lamina A, mostró un fenotipo progeroide más leve de lo habitual, posiblemente debido a la reducción de los niveles de prelamina A farnesilada72. El hecho de que los ratones Zmpste24-/- con haploinsuficiencia del gen LMNA no muestren un fenotipo de envejecimiento evidente apoya esta conclusión73. La importancia de la farnesilación en la patogénesis de la HGPS se ha confirmado gracias a la generación de ratones LmnacsmHG/csmHG, que producen progerina no farnesilada y no mueren prematuramente74. Asimismo, los ratones LmnanPLAO/nPLAO, que expresan únicamente prelamina A no farnesilada, desarrollan miocardiopatía, pero no progeria75. Además el tratamiento con inhibidores de la farnesil transferasa reduce las alteraciones nucleares observadas en las células que expresan progerina76,77 y previene el inicio y la progresión tardía de ECV en los ratones progeroides G608G BAC78, animales que contienen la versión del gen de la LMNA humana con la mutación c.1824C>T (p.G608G), que causa HGPS79. Análogamente, estos inhibidores prolongan la supervivencia de los ratones progeroides Zmpste24-/− y LmnaHG/+, caracterizados por expresar prelamina A y progerina, respectivamente79,80.
Trabajos posteriores mostraron que el tratamiento con un inhibidor de la farnesil transferasa provocaba la prenilación alternativa de la prelamina A y la progerina mediada por una geranilgeraniltransferasa81. El tratamiento combinado con estatinas y aminobisfosfonatos para bloquear tanto la farnesilación como la geranilgeranilación de la prelamina A mejoró el fenotipo progeroide y prolongó la vida de ratones82Zmpste24-/-. Con base en los hallazgos en células y ratones que expresan progerina y prelamina A11, se han desarrollado estudios clínicos en pacientes con HGPS para analizar el efecto del tratamiento con un inhibidor de la farnesil transferasa (lonafarnib) solo o combinado con estatinas (pravastatina) y bisfosfonatos (zoledronato)46,52,61. La monoterapia con lonafarnib proporcionó una cierta mejora sobre la rigidez vascular, la estructura ósea y el estado auditivo y se estimó que incrementa la supervivencia en 1,6 años46,61. El tratamiento basado en la combinación de 3fármacos (lonafarnib, pravastatina y zoledronato: triple drug trial) mostró una mejora adicional en la densidad ósea pero no hubo mejoría en los parámetros cardiovasculares en comparación con la monoterapia con lonafarnib52. Por tanto, aunque la progerina farnesilada parece jugar un papel fundamental en la HGPS, las terapias actuales dirigidas a prevenir la farnesilación de la progerina solo aportan un beneficio moderado.
La observación de que tanto la progerina farnesilada como no farnesilada se acumula en la membrana nuclear impulsó a Kalinowski et al. a proponer que la asociación de la progerina con la membrana nuclear interna también involucra el aumento en las interacciones electrostáticas y la agregación83. Además, las propiedades de la cola de la progerina, menos heterogénea y más compacta que la lamina A normal, podrían alterar su interacción con el ADN y con otras proteínas84.
Otro factor que podría contribuir a la toxicidad de la progerina es la alteración de su estructura proteica debido a la eliminación de 50 aminoácidos cerca de la región C-terminal. Las interacciones anormales de la progerina con otros componentes nucleares provocan la formación de protuberancias (blebbing) nucleares, un incremento del grosor y la rigidez de la lámina nuclear, la localización anómala de la heterocromatina y la alteración de los complejos de poro nuclear85,86. Recientemente, Lee et al.87 han descrito que la progerina interacciona fuertemente con la lamina A/C y que la disrupción química de los heterodímeros progerina-lamina A/C reduce las aberraciones nucleares, previene la senescencia celular, limita los rasgos progeroides y alarga la esperanza de vida en los ratones LmnaG609G/G609G.
La carboximetilación de la farnesilcisteína C-terminal de la progerina catalizada por la ICMT también podría contribuir a la progeria. La reducción de la expresión y actividad de la ICMT en un 70-90% en ratones hipomórficos Zmpste24-/-Icmthm/hm mejoró el peso corporal, la fuerza de agarre y la estructura ósea y alargó la supervivencia en comparación con ratones control Zmpste24-/-Icmt+/+con la ICMT intacta88. La disminución de la actividad ICMT en los ratones Zmpste24-/-Icmthm/hm se asoció con una alteración en la localización de la prelamina A y con la activación de la señalización mediada por AKT (proteína cinasa B, protein kinase B) y mTOR (mammalian target of rapamycin), lo que a su vez retrasó la senescencia celular. Sin embargo, conviene señalar que la rapamicina, inhibidora de mTOR, activó la eliminación de la progerina mediante autofagia y redujo las anormalidades nucleares89,90. Estos resultados muestran claramente que ICMT y mTOR están implicados en el envejecimiento prematuro, sin embargo, se requerirán nuevos estudios para definir de forma precisa los mecanismos implicados, y la relación entre ICMT, AKT y mTOR.
Mecanismos implicados en el desarrollo de enfermedad cardiovascular en la progeriaEsta sección recoge la información disponible hasta el momento sobre los mecanismos celulares y moleculares mediante los cuales la prelamina A y la progerina dañan el sistema cardiovascular. Este conocimiento es fundamental para comprender los mecanismos implicados en el desarrollo de ECV durante el envejecimiento normal, puesto que tanto la prelamina A como la progerina se expresan en niveles bajos en células y tejidos de individuos sanos no progéricos, incluyendo CMLV de la media y lesiones ateroscleróticas15,16,91.
Pérdida de células musculares lisas de la pared vascularLa pérdida progresiva de CMLV es una característica de los pacientes con HGPS15,53,92 y de los modelos de progeria en ratones66,67,79, lo que indica su contribución en la enfermedad vascular progeroide. Aunque menos grave, la reducción en el contenido de CMLV en la media también tiene lugar durante el envejecimiento fisiológico93.
Las CMLV están sujetas a un elevado estrés mecánico relacionado con el flujo sanguíneo. En condiciones normales las células responden al aumento del estrés de cizalladura (shear stress) aumentando la expresión de la lamina A/C y modificando su localización nuclear62,94,95. En células que expresan progerina las respuestas anómalas al estrés físico pueden conllevar daño y muerte celular86,96. En concordancia con esta idea, se ha descrito que el estrés mecánico aplicado a fibroblastos de pacientes con HGPS reduce la viabilidad e incrementa la muerte celular por apoptosis97. Las alteraciones en la mecanotransducción inducidas por la progerina podrían explicarse por los cambios en la expresión de proteínas que controlan la organización del citoesqueleto, la mecanotransducción y la producción de la matriz extracelular98,99. De hecho, la aorta ascendente de ratones transgénicos BAC G608G que expresan progerina presenta una disminución en la expresión de vimentina, una proteína del citoesqueleto que se une al núcleo, al retículo endoplásmico y a la mitocondria y que es esencial para el mantenimiento de la integridad celular100. Esta correlación entre reducción de la expresión de proteínas de mecanotransducción y aumento del estrés de cizalladura podría explicar parcialmente la pérdida de CMLV en HGPS.
Los mecanismos que subyacen a la pérdida de CMLV provocada por la progerina pueden analizarse utilizando protocolos de diferenciación a partir de células madre pluripotentes inducidas (induced pluripotent stem cells, iPSC) derivadas de individuos sanos y de pacientes con HGPS. Liu et al. observaron que las CMLV diferenciadas a partir de iPSC que expresan progerina presentan senescencia prematura como consecuencia de la interacción de la progerina con la subunidad catalítica de la proteína cinasa dependiente de ADN, una subunidad catalítica de la ADN-PK nuclear que participa en la reparación del ADN mediante recombinación no homóloga101. En cambio, Kinoshita et al. describieron que la progerina, a diferencia de la lamina A, no puede interaccionar con la ADN-PK o con otras proteínas implicadas en la respuesta al daño del ADN102. También observaron que la expresión de la progerina en CMLV, pero no en CE, induce la activación de la ADN-PK, lo que frenaría el crecimiento de las CMLV e induciría su senescencia. En vista de estas discrepancias, es evidente que se necesitarán nuevos estudios para aclarar la interacción entre la progerina y la ADN-PK y sus subunidades en diferentes tipos celulares y para establecer las consecuencias fisiopatológicas.
Zhang et al. describieron una proliferación defectuosa de las CMLV derivadas de iPSC de pacientes con HGPS, como consecuencia de mecanismos independientes de la activación de caspasas103. Estos autores también observaron que la expresión de la progerina en CMLV inhibe la poli(ADP-ribosa)-polimerasa 1, un regulador clave de la reparación del ADN, y que activa la respuesta de la recombinación no homóloga propensa a errores, lo que prolonga la mitosis y provoca catástrofe mitótica y muerte celular. La prelamina A también induce daño en el ADN e incrementa la respuesta celular al daño del ADN en CMLV envejecidas16,104. Esta respuesta podría ser consecuencia de un reclutamiento defectuoso de la proteína 1 de unión a p53 (53BP1) a regiones dañadas del ADN, lo que perturbaría el transporte al núcleo debido a una alteración de la localización105 de la nucleoporina 153. La alteración de la reparación del ADN dañado también se ha descrito en células no vasculares procedentes de pacientes con HGPS y en modelos de progeria en ratones106-108. Todos estos hallazgos confirman que la muerte de CMLV inducida por progerina se debe, al menos en parte, a defectos en la reparación del ADN, una alteración que también juega un papel fundamental en el envejecimiento normal109.
Calcificación vascularAl igual que los pacientes con HGPS15,55-57, los ratones progeroides G608G BAC y LmnaG609G/+ desarrollan calcificación aórtica79,110. Las aortas calcificadas de los ratones LmnaG609G/+ mostraron una expresión anormalmente elevada de marcadores osteogénicos, entre ellos la proteína morfogénica ósea 2 (bone morphogenetic protein, Bmp2) y el factor de transcripción 2 relacionado con Runt (Runt-related transcription factor 2, Runx2), sin que existiera alteración en agentes anticalcificantes, como la proteína Matrix-Gla y la fetuina A110. Asimismo, CMLV primarias derivadas de ratones LmnaG609G/+ mostraron menor capacidad para inhibir la deposición de calcio in vitro, lo que se asoció con una menor concentración extracelular de pirofosfato inorgánico (ePPi), el principal inhibidor endógeno de la calcificación vascular. La reducción de los niveles de ePPi en CMLV en cultivo estaba relacionada con una reducción de su síntesis causada por la disminución de la producción de ATP (el principal sustrato en la síntesis de ePPi) y la inducción tanto de la fosfatasa alcalina no específica de tejido (tissue-nonspecific alkaline phosphatase, la enzima fundamental en la hidrólisis del PPi), como de la ectonucleósido trifosfatasa difosfohidrolasa 1 (ectonucleoside triphosphatase diphosphohydrolase 1, una enzima que hidroliza ATP para liberar PPi). De hecho, los ratones LmnaG609G/+ mostraron concentraciones plasmáticas de ePPi y ATP inferiores a las de sus hermanos de camada Lmna+/+, mientras que el tratamiento con PPi exógeno previno la calcificación vascular en los ratones LmnaG609G/G609G110.
La expresión de la prelamina A en CMLV promueve la calcificación vascular mediante la activación de la ataxia/telangiectasia mutada/ataxia telangiectasia relacionada con Rad3104, una vía de señalización asociada con el daño del ADN. La activación de esta vía en CMLV induce el fenotipo secretor asociado a la senescencia celular, con liberación de factores procalcificantes tales como BMP2 que pueden desencadenar la calcificación tanto local como en regiones remotas104. Además, el cultivo de CMLV en un medio procalcificante conlleva la inducción de la expresión de la lamina A y la prelamina A, junto con un incremento de la expresión de factores procalcificantes como Runx2, osteocalcina y osteopontina y un aumento de la deposición de calcio111. Hay que destacar que células madre mesenquimales humanas que expresan progerina también presentan niveles elevados de osteopontina y muestran un aumento de la diferenciación osteogénica112.
En resumen, la lamina A y sus formas mutadas o no procesadas participan en la diferenciación osteoblástica y en la calcificación de las CMLV, lo que pone de manifiesto la necesidad de profundizar en nuestro conocimiento acerca del papel de la progerina y la prelamina A en la calcificación vascular durante el envejecimiento prematuro.
Disfunción endotelialLa disfunción endotelial juega un papel fundamental en todas las fases de la aterosclerosis, la enfermedad que compromete la vida de los pacientes con HGPS. Las CE detectan y responden a diferentes tipos de flujo sanguíneo y las regiones aórticas sometidas a un flujo sanguíneo turbulento y a un elevado estrés de cizalladura, tales como la aorta ascendente, son más susceptibles al desarrollo de aterosclerosis113,114. Song et al. detectaron monocapas de CE intactas en zonas de la aorta ascendente de ratones G608G BAC prácticamente sin CMLV98. Las CE que expresan progerina próximas a regiones que han sufrido una pérdida masiva de CMLV presentan una expresión de vimentina más de 8 veces superior a las de las CE localizadas en regiones que conservan su contenido de CMLV, lo que las hace más resistentes al estrés de cizalladura y puede explicar la presencia de un endotelio bien preservado en vasos de pacientes con HGPS15.
La expresión elevada de moléculas de adhesión en las CE disfuncionales desencadena la adhesión de los monocitos, una etapa importante en el inicio y la progresión de la aterosclerosis31. Estudios recientes muestran que la acumulación de prelamina A en CE debida al bloqueo de la maduración de la lamina A induce senescencia celular y promueve la adhesión de monocitos de forma dependiente de la molécula de adhesión intercelular 1 (intercellular adhesion molecule 1)115. Es evidente que la caracterización de la conexión entre las CE, la expresión de progerina y prelamina A y la progresión de la placa de ateroma requerirán nuevos estudios.
Conclusiones finales y perspectivasEl envejecimiento es el principal factor de riesgo de ECV. Ante el envejecimiento progresivo de nuestra sociedad y puesto que la ECV constituye la principal causa de morbimortalidad a nivel mundial, es urgente mejorar nuestro conocimiento acerca de los mecanismos que contribuyen al envejecimiento. Esta información es fundamental para el desarrollo de nuevas estrategias que reduzcan la aparición y el desarrollo de enfermedad en la vejez y, por tanto, para promover un envejecimiento saludable. La intensa labor realizada en el ámbito de la investigación básica, clínica y epidemiológica ha permitido identificar los mecanismos generales implicados en el envejecimiento. El reto en la investigación sobre envejecimiento es identificar cuáles de estos mecanismos contribuyen principalmente a explicar la elevada variabilidad interindividual en el envejecimiento biológico humano. Este conocimiento contribuirá al desarrollo de nuevas terapias y a mejorar la prevención mediante la identificación temprana de aquellos individuos que presenten un mayor riesgo de sufrir enfermedades relacionadas con la edad, antes de que aparezcan los primeros síntomas. Estas actuaciones promoverán una vejez saludable y reducirán el impacto socioeconómico y sobre los sistemas de salud que provoca el envejecimiento.
El envejecimiento y la ECV están fuertemente acelerados en pacientes con HGPS, un desorden genético muy raro causado por la progerina, una forma no procesada de la lamina A. La progeria en humanos también se asocia con la acumulación anormal de la prelamina A debido a mutaciones que inactivan la proteasa ZMPSTE24. Es importante destacar que tanto la prelamina A como la progerina se expresan, aunque a niveles bajos, en células y tejidos de ancianos, incluidas las células de la pared vascular. En células de individuos sin HGPS se ha propuesto que el estrés oxidativo y el acortamiento de los telómeros inducen la expresión de la prelamina A y la progerina, respectivamente. La investigación en progeria puede, por tanto, ayudar a identificar los mecanismos celulares y moleculares que dirigen el envejecimiento normal y la ECV asociada. Los factores de riesgo cardiovascular tradicionales tales como la hipercolesterolemia, la diabetes, la obesidad, la hipertensión y el tabaquismo suelen estar ausentes, o ser leves, en los pacientes con HGPS. Por tanto, la investigación en esta enfermedad ofrece una oportunidad única para diferenciar aquellos mecanismos que inducen daño cardiovascular de forma directamente dependiente de la edad de aquellos factores de riesgo modificables que progresivamente deterioran las células y tejidos durante la vejez y que pueden afectar a la salud cardiovascular de forma secundaria.
La identificación de los mecanismos, tanto específicos como comunes, implicados en el desarrollo del envejecimiento normal y prematuro requerirá estudios genómicos, epigenómicos, transcriptómicos, proteómicos y metabolómicos a gran escala. Asimismo, se podrán establecer relaciones causales mediante estrategias de pérdida y ganancia de función dirigidas contra los factores candidatos identificados en los estudios «ómicos». Considerando el gran número de tipos celulares que participan en la ECV y en el envejecimiento normal y prematuro, será fundamental la generación de nuevos modelos animales condicionales o específicos de tejido, con especial énfasis en aquellas células que juegan un papel crítico en la aterosclerosis (ej. monocitos/macrófagos, linfocitos, CE y CMLV). Puesto que los modelos animales de progeria generados hasta el momento no desarrollan aterosclerosis, la principal causa de muerte en pacientes con HGPS, será fundamental la generación de modelos de progeria que presenten aterosclerosis, tanto en animales de pequeño como de gran tamaño. Los retos del futuro estarán centrados en establecer cómo el envejecimiento fisiológico conduce a la acumulación de formas no procesadas de la lamina A y en determinar si las anomalías nucleares inducidas por estas proteínas contribuyen al envejecimiento normal.
FinanciaciónEl trabajo desarrollado en el laboratorio de V. A. se ha financiado por el Ministerio de Economía, Industria y Competitividad (MEIC; SAF2016-79490-R) y el Instituto de Salud Carlos III (ISCIII) (RD12/0042/0028 y AC16/00091), con cofinanciación del Fondo Europeo de Desarrollo Regional (FEDER), de la Fundació Marató de TV3 (122/C/2015) y la Progeria Research Fundation (Established Investigator Award 2014-52). El CIBER de Enfermedades Cardiovasculares es una iniciativa del ISCIII.
L. C. recibió una ayuda del programa de becas posdoctorales Jordi Soler de la Red de Investigación Cardiovascular (ISCIII) y M. R. H., una ayuda predoctoral del programa FPI del MEIC (BES-2011-043938). El CNIC cuenta con el apoyo del MEIC y de la Fundación Pro-CNIC y ha sido acreditado como Centro de Excelencia Severo Ochoa (MEIC SEV-2015-0505).
Los autores piden disculpas a todos aquellos investigadores cuyo trabajo no ha podido ser citado debido a las limitaciones de espacio.