





La aterosclerosis es la enfermedad cardiovascular con más morbimortalidad en países desarrollados. Los nuevos hábitos de vida, la falta de mecanismos eficaces en la detección temprana de la enfermedad y la escasez de tratamientos farmacológicos específicos pueden ser la causa de este fenómeno. Un episodio clave en la formación de la placa aterosclerótica es el componente inflamatorio. La búsqueda de nuevos mediadores inflamatorios en la enfermedad podría mostrar nuevas dianas para diseñar tratamientos farmacológicos en el futuro. Entre otros, actualmente se están estudiando las citocinas de la familia del factor de necrosis tumoral, las moléculas proinflamatorias prostanoides y los agentes protectores como HSP (del inglés heat shock proteins).
Atherosclerosis is the cardiovascular disease with highest morbidity-mortality in developed countries. New life habits, lack of efficient mechanisms in the early detection of the disease and the limited specific pharmacological treatments, may be the cause of this phenomenon. A key event in the formation of the atherosclerotic plaque is the inflammatory component. The search for new inflammatory mediators in this disease could show new targets to design future pharmacological treatments. Currently being studied are, among others, the cytokines of the TNF (Tumour Necrosis Factor) family, the prostanoid pro-inflammatory molecules and protector agents, such as the HSPs (Heat Shock Proteins).
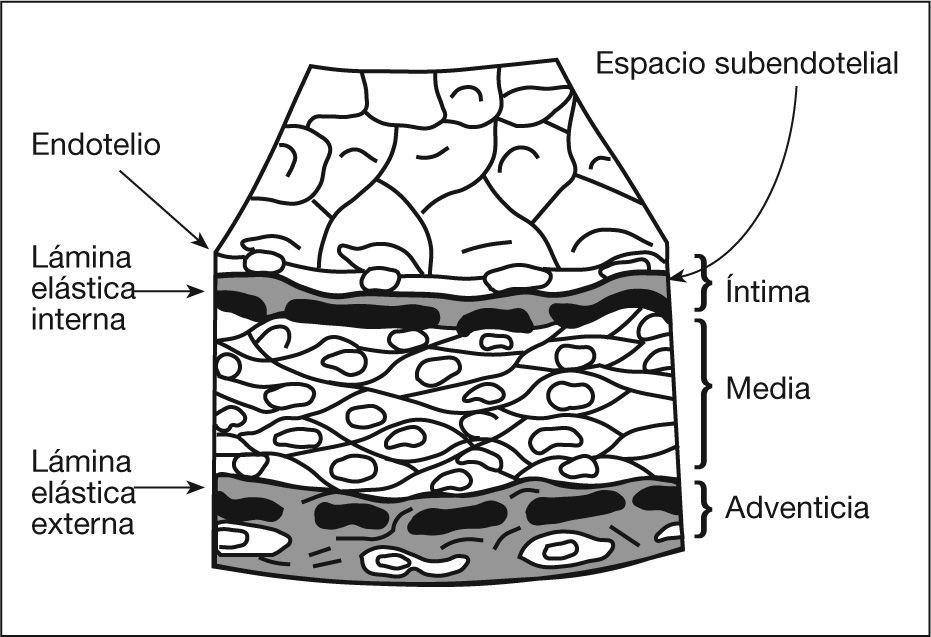
La pared arterial consta de 3 capas: íntima, media y adventicia. La íntima está formada por células endoteliales situadas sobre una lámina elástica interna (fig. 1). El endotelio, además de servir como barrera limitante para la sangre, regula el intercambio molecular entre ésta y el interior de la pared vascular. La capa media está compuesta por células musculares lisas (CML), que son las encargadas del tono vascular de la pared. La permeabilidad endotelial y el grado de relajación-contracción muscular dependen de la producción celular de óxido nítrico (NO) y prostaglandinas. La capa adventicia está separada de la media por la lámina elástica externa, y en ella se ubican las terminaciones nerviosas y la red de microvasos del vasa vasorum. Las capas íntima y media serán las principales capas afectadas por la enfermedad aterosclerótica.

Disposición de las diferentes capas en la pared vascular. Desde la luz del vaso, la pared vascular está compuesta por íntima, de la que forma parte el endotelio, y que es el lugar donde se inicia la formación de la placa de ateroma. Hacia fuera se aprecia la capa muscular media y, más externamente, la adventicia.
La aterosclerosis representa la causa principal de muerte en países desarrollados. Debido a los nuevos estilos de vida (dietas, sedentarismo), comienza a afectar a países en desarrollo. Se caracteriza por el engrosamiento de la pared arterial de vasos grandes y medianos (en zonas de rozamiento menor con el flujo sanguíneo), debido a la formación de la placa de ateroma. La formación de la placa provoca el estrechamiento del lumen del vaso y, por tanto, la reducción del aporte sanguíneo a órganos como el corazón y el cerebro. La placa aterosclerótica puede presentar una rotura y causar un coágulo sanguíneo que ocluye la circulación, y provocar un episodio vascular isquémico agudo. El fenómeno de la aterogenia es un proceso complejo, uno de cuyos principales componentes es el proceso inflamatorio. La selección celular, la proliferación, la neovascularización y la esclerosis son episodios asociados a la iniciación y la evolución de la placa. Actualmente, las 2 estrategias terapéuticas principales se basan en la manipulación del metabolismo de lipoproteínas plasmáticas o colesterol celular y en la reducción de los procesos inflamatorios asociados.
Desarrollo y evolución de la placa de ateromaLa enfermedad aterosclerótica ocurre con más frecuencia en las regiones del árbol vascular, donde se altera el flujo laminar sanguíneo. La disfunción endotelial que inicia la aterosclerosis puede derivarse de numerosos factores, como dislipemia, hipertensión arterial, hiperglucemia, incremento de la producción de radicales de oxígeno (ROS) o activación del sistema simpático-adrenal. El NO es la causa principal de la regulación de la permeabilidad. En esta enfermedad se produce una disminución de la disponibilidad de NO. Durante el desarrollo de la placa, otros componentes de la estría grasa provendrán de capas más externas del vaso.
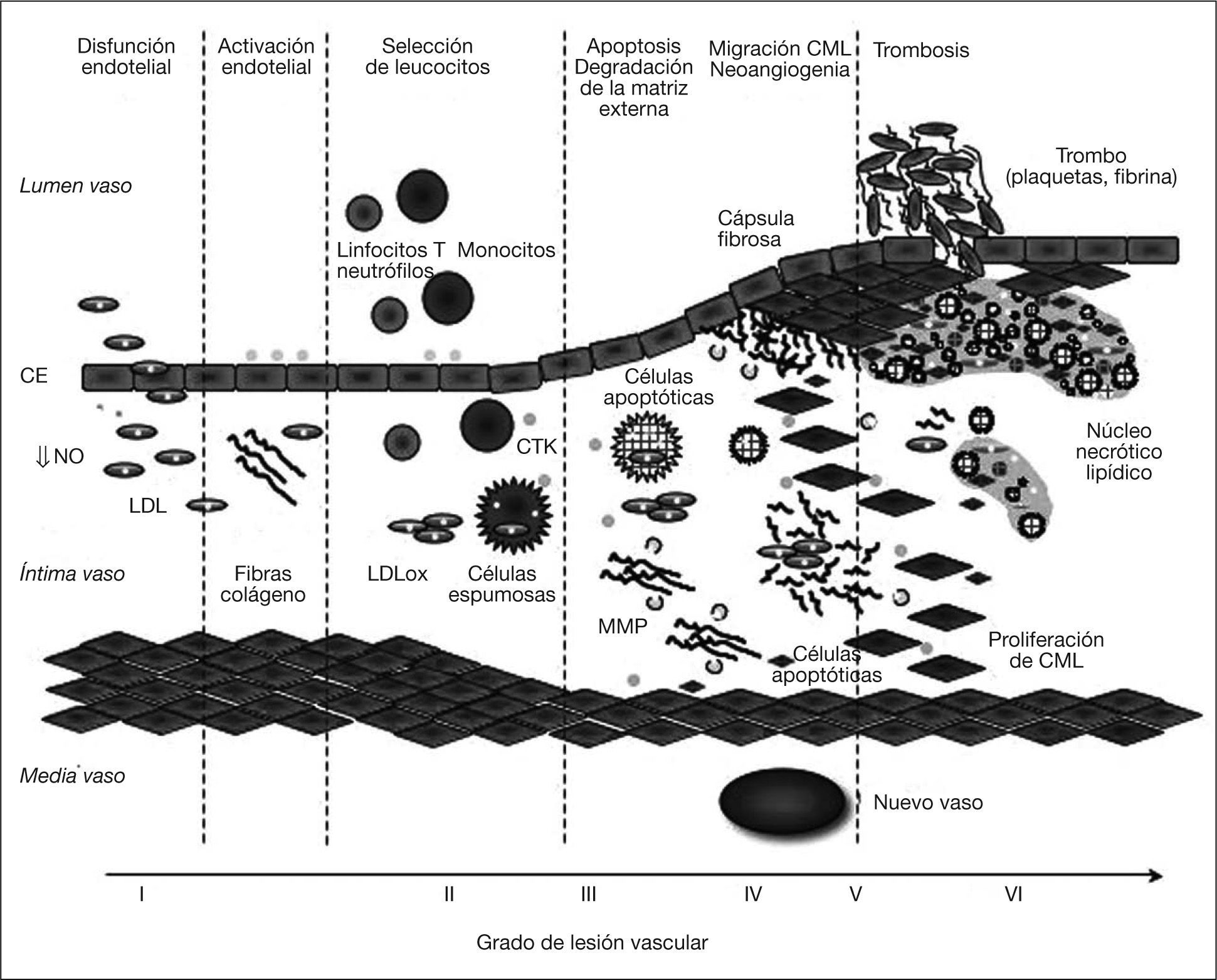
Etapas de la aterogeniaLas lipoproteínas aterogénicas circulantes, como las lipoproteínas de baja densidad (LDL) transendotelizan hacia la íntima cuando aumenta la permeabilidad de la pared debido a la disfunción endotelial. Una vez allí, las LDL se oxidan o modifican enzimáticamente (glucosiladas), por lo que sus receptores celulares no pueden reconocerlas y se acumulan y agregan en el espacio extracelular. Este fenómeno hace desencadenar una respuesta inflamatoria que atrae leucocitos, principalmente linfocitos y monocitos, desde el torrente sanguíneo. Estas células se adhieren al endotelio gracias a las moléculas de adhesión, y penetran hacia el espacio intimal por acción de diversas quimiocinas1,2. La adhesión de los leucocitos al endotelio requiere también la reducción de los valores de NO. La selección de linfocitos T perpetúa el proceso de inflamación hasta hacerlo crónico. En la íntima, los monocitos se diferencian a macrófagos para fagocitar las lipoproteínas y transformarse en células espumosas. La acumulación de células espumosas comienza la formación de la estría grasa (fig. 2).
![Evolución de la placa de aterosclerosis. Iniciación de la lesión (disfunción endotelial y producción de moléculas de adhesión); progresión (infiltración de lipoproteínas y atracción de monocitos); crecimiento (acumulación de células espumosas y liberación de moléculas proinflamatorias/apoptóticas); esclerosis (migración de células musculares lisas [CML] y formación de capa fibrosa); desestabilización (apoptosis de CML y macrófagos, producción de proteasas y angiogenia) y rotura de la placa y trombosis. CE, CTK y MMP corresponden a células endoteliales, citocinas/quimiocinas y metaloproteinasas, respectivamente. LDL: lipoproteínas de baja densidad; NO: óxido nítrico.](https://static.elsevier.es/multimedia/02149168/0000002100000001/v1_201305021840/S0214916809702785/v1_201305021840/es/main.assets/gr2.jpeg?xkr=ue/ImdikoIMrsJoerZ+w997EogCnBdOOD93cPFbanNcxT+B4FjbiPdoCJto3d5S/Ch6vvG+Cp80/Jya7nfY0UubDf2JcIC1PG/BqUF42x7WhHuWxt2qEs3wbG/AfH+i3ICC1Pfd0v2boiekXjM32/G4zyBHnYVk/nDW3q1bhB7qZTihjWiGUWPj+K5lYh4ouBxB4CESPvLsnBtUSc88YUwHjXGaqLDp8SSCAhvICBT36Px06zDMlKs/mzIwoxIyjorb0jMXVznAi1Z6lk+jeT0sTxiBGZLuGLz2GqeBLLnc= "Evolución de la placa de aterosclerosis. Iniciación de la lesión (disfunción endotelial y producción de moléculas de adhesión); progresión (infiltración de lipoproteínas y atracción de monocitos); crecimiento (acumulación de células espumosas y liberación de moléculas proinflamatorias/apoptóticas); esclerosis (migración de células musculares lisas [CML] y formación de capa fibrosa); desestabilización (apoptosis de CML y macrófagos, producción de proteasas y angiogenia) y rotura de la placa y trombosis. CE, CTK y MMP corresponden a células endoteliales, citocinas/quimiocinas y metaloproteinasas, respectivamente. LDL: lipoproteínas de baja densidad; NO: óxido nítrico.")
Evolución de la placa de aterosclerosis. Iniciación de la lesión (disfunción endotelial y producción de moléculas de adhesión); progresión (infiltración de lipoproteínas y atracción de monocitos); crecimiento (acumulación de células espumosas y liberación de moléculas proinflamatorias/apoptóticas); esclerosis (migración de células musculares lisas [CML] y formación de capa fibrosa); desestabilización (apoptosis de CML y macrófagos, producción de proteasas y angiogenia) y rotura de la placa y trombosis. CE, CTK y MMP corresponden a células endoteliales, citocinas/quimiocinas y metaloproteinasas, respectivamente. LDL: lipoproteínas de baja densidad; NO: óxido nítrico.
La respuesta inflamatoria atrae más células sanguíneas y hace migrar además CML desde la capa media hacia la íntima del vaso, donde proliferan y secretan componentes de matriz extracelular (colágenos), para formar la cápsula fibrosa (fig. 2). Esta cápsula da consistencia y estabilidad a la placa aterosclerótica (lesión esclerótica). Sin embargo, la presencia de CML incrementa la retención y la agregación de lipoproteínas aterogénicas. En las primeras fases de crecimiento de la placa se produce un remodelado de la pared vascular, con el aumento del diámetro total del vaso. De este modo, la luz vascular conserva así su tamaño al compensarse la obstrucción producida por la lesión aterosclerótica. La muerte de los macrófagos hace liberar su contenido (LDL, ROS) al espacio intracelular de la íntima, con lo que se crea un núcleo necrótico tóxico que promueve la selección de más células inflamatorias y CML, y la liberación de proteasas (metaloproteinasas). Estas enzimas van a degradar las proteínas de matriz de la cápsula fibrosa haciéndola más vulnerable (fig. 2). La estabilidad de la placa depende en mayor medida de la integridad de la capa fibrosa y de sus componentes de matriz extracelular. Hay zonas con más acumulación de macrófagos, inflamación y muerte celular localizadas en los hombros de la placa1–3.
Otro fenómeno importante en la evolución de la placa es la formación de microvasos debido a la liberación de moléculas angiogénicas, como el factor de crecimiento vascular del endotelio (VEGF). La neoangiogenia contribuye a la aterogenia al aumentar la superficie de contacto con la sangre, lo que permite atraer más células inflamatorias, e incrementar la probabilidad de hemorragias. Recientemente, se ha indicado que el colesterol de las membranas de los hematíes así depositados en el interior de la placa podría contribuir a su desestabilización4.
Finalmente, si la placa se rompe, expondrá el material trombogénico hacia el lumen del vaso y favorecerá la adhesión de plaquetas para formar el trombo (fig. 2). El trombo podrá ocluir de forma total o parcial la luz del vaso o liberarse y bloquear en áreas de menor calibre, causando un síndrome coronario agudo, ictus o isquemia periférica5.
Factores moleculares implicados en la progresión de la placaEn la respuesta inflamatoria del proceso aterosclerótico, la selección de leucocitos está regulada por moléculas de adhesión y NO expresadas por células endoteliales. Estas células producen además citocinas y quimiocinas, que son la causa de la diferenciación de monocitos y de la migración y proliferación de las CML en la capa media del vaso. Las CML liberan más citocinas y factores proinflamatorios (prostaglandinas), con lo que así se perpetúa el daño. La regulación de la producción y la degradación de la matriz extracelular por las citocinas y proteasas desempeñará un papel decisivo en el destino de la placa. Debido a la importancia de los procesos inflamatorios en la enfermedad aterosclerótica, el estudio de nuevos mediadores proinflamatorios puede proporcionar dianas terapéuticas frente a las que diseñar fármacos para el tratamiento de esta enfermedad. Entre estos nuevos mediadores, tiene especial relevancia el sistema TWEAK/factor.
Papel del sistema TWEAK/Fn14 en la aterosclerosisLa superfamilia de citocinas proinflamatorias factor de necrosis tumoral (TNF) puede desempeñar un papel clave en la formación y la progresión de la placa6. El TNF se libera desde las células endoteliales, leucocitos y CML como respuesta a agentes tóxicos o proinflamatorios. El TNF inductor débil de apoptosis (TWEAK) es un miembro de la familia de TNF. TWEAK es inicialmente una proteína transmembrana tipo II cuyo extremo N-terminal hidrofóbico se dirige hacia el citosol celular7. El dominio C-terminal es extracelular y, al igual que otros miembros de la familia de TNF, contiene una secuencia de corte para furina. TWEAK se procesa de forma rápida por la furina para dar lugar a la forma soluble y activa de la proteína. El receptor o los receptores celulares para TWEAK aún se están estudiando. Utilizando librerías de cADN humanas se ha descubierto un receptor para TWEAK llamado factor de crecimiento 14 inducible de fibroblastos (Fn14). Fn14 es una proteína transmembrana tipo I que es sintetizada con un péptido señal en su región N-terminal, el cual se escinde para dar lugar a la proteína madura Fn14. Recientemente, se ha demostrado que TWEAK no interactúa con otros miembros de la familia de TNF. Sin embargo, se ha indicado que los efectos biológicos de TWEAK podrían ser mediados por receptores diferentes de Fn14 en monocitos/macrófagos8,9. Esta citocina se expresa en páncreas, intestino, corazón, cerebro, pulmón, ovario, músculo esquelético y, en menor medida, en hígado y riñón. Linfocitos, macrófagos, células endoteliales, mesangiales y CML son capaces de expresar TWEAK. Fn14 se expresa en corazón, pulmón, riñón y placenta, además de en células endoteliales, monocitos/macrófagos, célulasmesangiales y CML. En placas ateroscleróticas, ambas proteínas se expresan tanto por CML como por macrófagos.
En aterosclerosis, la interacción de TWEAK con Fn14 puede desencadenar varias respuestas biológicas durante la formación de la placa aterosclerótica, como estimulación de la proliferación, migración, apoptosis, angiogenia y producción de otras citocinas proinflamatorias (fig. 3). Todos estos episodios participan en los diferentes estadios del desarrollo de la placa7–9.
 antes de unirse al receptor Fn14 (u otro aún no descrito). La activación de Fn14 atrae TRAF (del inglés TNF, receptor-associated factor) a la membrana y transactiva proteínas intracelulares MAP-cinasas (MAPC) y/o factor nuclear de transcripción kappa B (NF-κB). Como consecuencia, se estimula la transcripción de genes proapoptóticos, inflamatorios, angiogénicos, proliferativos y, posiblemente, de degradación de matriz extracelular.")
Señalización del sistema TWEAK-Fn14. TWEAK oligomeriza (3 monómeros) antes de unirse al receptor Fn14 (u otro aún no descrito). La activación de Fn14 atrae TRAF (del inglés TNF, receptor-associated factor) a la membrana y transactiva proteínas intracelulares MAP-cinasas (MAPC) y/o factor nuclear de transcripción kappa B (NF-κB). Como consecuencia, se estimula la transcripción de genes proapoptóticos, inflamatorios, angiogénicos, proliferativos y, posiblemente, de degradación de matriz extracelular.
Respecto a la iniciación y la progresión de la lesión, se ha descrito que el endotelio sano no es capaz de unir e internalizar monocitos, debido en parte a la acción del sistema Fas (otros miembros de la familia TNF). El ligando de Fas induce la apoptosis de los monocitos que intentan invadir la pared vascular en ausencia de estímulos proinflamatorios. En condiciones patológicas, las células endoteliales disminuyen la expresión de ligando de Fas y aumentan la de moléculas de adhesión en superficie, como ICAM (del inglés intercellular adhesion molecule), selectinas y VCAM (del inglés vascular cell adhesion molecule), que actúan como receptores para proteínas de superficie de leucocitos. TWEAK es capaz de estimular la expresión de ICAM-1, E-selectina y citocinas/quimiocinas proinflamatorias, como la interleucina (IL) 8 y MCP-1. Estas moléculas atraerán monocitos al foco inflamatorio (efecto proaterogénico del sistema TWEAK/Fn14).
El papel de TWEAK en la desestabilización y la formación de trombo se ha relacionado con su capacidad de inducir angiogenia, así como proliferación y migración de diferentes células vasculares como las CML. Además, se ha detectado expresión de TWEAK y Fn14 junto a metaloproteinasas en regiones de la placa ricas en células espumosas, por lo que se ha indicado su implicación en la degradación de la capa fibrosa y desestabilización de la placa. Además, TWEAK tiene propiedades proapoptóticas en determinadas situaciones donde coexisten otros factores proaterogénicos, como interferón gamma (IFN-γ). En monocitos estimulados con IFN-γ, se ha detectado una expresión alta de TWEAK. Es posible que las CML migrantes sean la diana del TWEAK liberado por los monocitos en condiciones proinflamatorias. El mecanismo de apoptosis inducido por esta molécula no se ha descrito completamente, aunque se ha demostrado que activa a las caspasas 8 y 3, y otras vías apoptóticas, como la de la catepsina B.
TWEAK y factor proinflamatorio NF-κBLa expresión de citocinas, moléculas de adhesión y factores protrombóticos está vinculada a la activación del factor nuclear de transcripción kappa B (NF-κB)10. Este factor, sensible al balance redox del microambiente, es capaz de inducir la transcripción de numerosos genes en multitud de tipos celulares (macrófagos, células endoteliales y vasculares). NF-κB desempeña un papel fundamental en el desarrollo de la placa aterosclerótica, al estar implicado en la regulación de NO, en la síntesis de lipoproteínas, citocinas/quimiocinas, metaloproteinasas y prostaglandinas, en la activación de moléculas de adhesión y factor tisular y en numerosas respuestas del sistema renina-angiotensina7–10. Hay una gran variedad de estímulos como LDL, angiotensina y citocinas como TNF e interleucinas que pueden activar NF-κB. Una vez estimulado, Fn14 atrae moléculas adaptadoras citoplasmáticas, como las proteínas TRAF (del inglés TNF, receptor-associated factor), que están implicadas en la activación de diferentes vías intracelulares, como NF-κB, p38 y JNK/ERK cinasas (fig. 3). TWEAK puede comportarse como una citocina multifuncional. Se ha demostrado en varios tipos celulares que la asociación TWEAK/Fn14 es capaz de activar NF-κB (a través de la fosforilación de I-κB) e inducir la expresión de IL-6, IL-8, RANTES, MCP-1 e ICAM-1, todos ellos implicados en la evolución de la placa8. Sin embargo, se ha observado que TWEAK puede equilibrar su efecto proaterogénico debido a su capacidad de inhibir la activación del factor de transcripción STAT-1 e inducir la asociación de NF-κB a la histona deacitilasa 1, reprimiendo así la producción de citocinas11.
TWEAK como biomarcador de aterosclerosisLa forma soluble de TWEAK (sTWEAK) se encuentra en plasma y sus valores son fácilmente detectables. Mediante análisis diferencial de expresión proteica por técnicas proteómicas, se ha descrito que sTWEAK es secretado en menor medida por vasos con placas ateroscleróticas en comparación con vasos sanos. Las concentraciones plasmáticas de sTWEAK se encuentran disminuidas en individuos con aterosclerosis carotídea en comparación con individuos sanos. En pacientes asintomáticos, la concentración de sTWEAK circulante se correlaciona negativamente con el grosor íntima-media12. Además, en enfermedades asociadas a aterosclerosis, como la diabetes mellitus tipo 2 o en casos de daño renal avanzado (hemodiálisis crónica), los valores circulantes de sTWEAK están también reducidos10. La síntesis de TWEAK podría reducirse en aterosclerosis como mecanismo de respuesta para evitar su efecto proaterogénico (expresión de moléculas de adhesión, citocinas/quimiocinas y metaloproteinasas) y proapoptótico (CML). La disminución de los valores circulantes de sTWEAK podría servir como potencial biomarcador de aterosclerosis y enfermedades asociadas.
Modulación terapéutica de TWEAK en la aterosclerosisLas estatinas se utilizan de forma asidua en el tratamiento de la aterotrombosis. Estos fármacos inhiben la enzima HMG-CoA reductasa, así como la biosíntesis de colesterol y, consecuentemente, de LDL. En enfermedades cardiovasculares, las estatinas tienen además efectos beneficiosos al bloquear la isoprenilación de proteínas G pequeñas (Ras, Rac, Rho) por intermediarios de la ruta del colesterol (mevalonato). Recientemente, se ha estudiado la posible relación entre el efecto de las estatinas y el sistema TWEAK/Fn147,12. En CML se ha observado que las estatinas pueden reducir la expresión de Fn14 inducida por citocinas. La adición de mevalonato revierte este proceso, lo que indica que los metabolitos de la ruta del colesterol son esenciales para la síntesis del receptor de TWEAK. Además, en estas células, el tratamiento con estatinas disminuye la respuesta inflamatoria inducida por TWEAK. De modo similar, estos fármacos reducen la expresión de Fn14 en placas carótidas humanas.
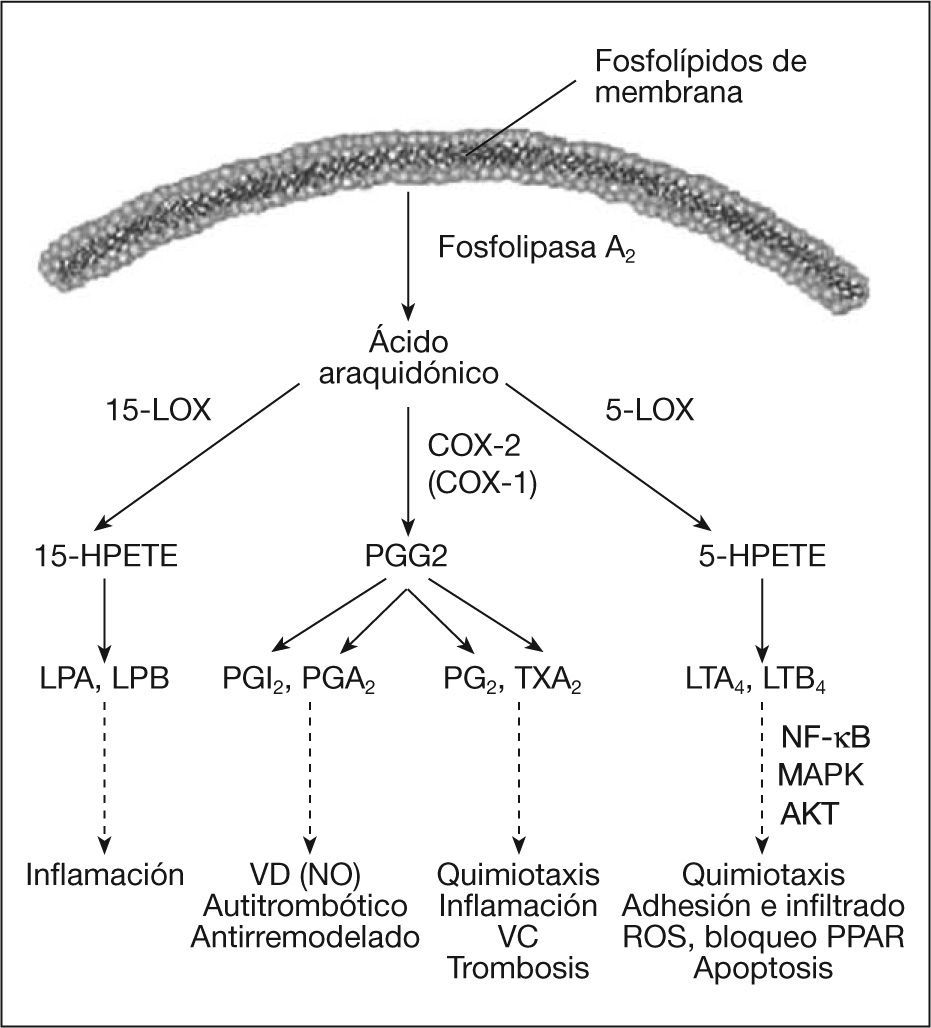
Papel de los leucotrienos y prostanoides en la aterosclerosisLos leucotrienos son mediadores lipídicos inflamatorios derivados de la vía de la 5 lipooxigenasa (5-LOX) a partir del precursor ácido araquidónico (fig. 4). El leucotrieno 4 (LTB-4) es uno de los productos finales y tiene propiedades quimioatractantes y proinflamatorias en numerosas enfermedades, incluida la aterosclerosis. La expresión de LTB-4 y todos los metabolitos necesarios para su biosíntesis está aumentada en placas ateroscleróticas y en plasma de pacientes con esta enfermedad13. LTB-4 se une a 2 receptores de membrana, BLT-1 y BLT-2, que se expresan en células sanguíneas y vasculares; además, media la acumulación y el filtrado de lípidos y leucocitos en la pared vascular, y promueve la apoptosis y la proliferación celular. Este leucotrieno es clave en la regulación de la quimiotaxis, la granulación, la fagocitosis y la generación de radicales superóxido en neutrófilos. LTB-4 puede inhibir a los receptores nucleares PPAR encargados de la proliferación de peroxisomas y del control del estrés oxidativo14. También promueve la conversión de células monocíticas a espumosas a través de la expresión del receptor CD36 para lipoproteínas y posterior acumulación de ácidos grasos. Por todo ello, LTB-4 está altamente implicado en la progresión y la rotura de las placas ateroscleróticas. Utilizando antagonistas del receptor BLT-1, el de mayor afinidad para LTB-4, se observa una reducción de la acumulación de lípidos y del infiltrado de monocitos en placas de aorta (en ratones knockout para la apolipoproteína [apo] E y el receptor de las LDL). En otros estudios, el empleo de estos antagonistas disminuyó los valores de moléculas de adhesión CD11b15–17.
 se sintetizan las diferentes prostaglandinas (PGI2, PGE2, TXA2) y leucotrienos (LTB4) gracias a las enzimas ciclooxigenasa (COX) y 5 lipooxigenasa (5-LOX), respectivamente. Las prostaglandinas y los leucotrienos están implicados de modo diferente en el desarrollo de la lesión aterosclerótica. El factor nuclear de transcripción kappa B (NF-κB) participa en la síntesis de estos mediadores mediante sus enzimas COX-2 y 5-LOX. HPETE: ácido hidroperoxioctadecadienoico; LPA: lipoxeno A; LPB: lipoxeno B; MAPK: MAP cinasas; VC: vasoconstricción; VD: vasodilatación.")
Metabolismo de las prostaglandinas y leucotrienos. Desde un mismo precursor (ácido araquidónico) se sintetizan las diferentes prostaglandinas (PGI2, PGE2, TXA2) y leucotrienos (LTB4) gracias a las enzimas ciclooxigenasa (COX) y 5 lipooxigenasa (5-LOX), respectivamente. Las prostaglandinas y los leucotrienos están implicados de modo diferente en el desarrollo de la lesión aterosclerótica. El factor nuclear de transcripción kappa B (NF-κB) participa en la síntesis de estos mediadores mediante sus enzimas COX-2 y 5-LOX. HPETE: ácido hidroperoxioctadecadienoico; LPA: lipoxeno A; LPB: lipoxeno B; MAPK: MAP cinasas; VC: vasoconstricción; VD: vasodilatación.
Los mecanismos intracelulares vinculados a la estimulación por LTB-4 son diversos. Se ha descrito que, a través de ambos receptores, LTB-4 puede activar proteínas ERK1/2, JNK1/2, p38, AKT y NF-κB. En células dominante-negativo para I-κKB (cinasa encargada de la activación de NF-κB), el receptor BLT-1 está anulado. A través de estas vías de señalización, LTB-4 aumenta la expresión de genes proinflamatorios, como IL-6, MCP-1 y TNF-α. Además, en un modelo de daño vascular, la inhibición de 5-LOX suprimió la migración de CML y redujo el tamaño neointimal18,19. Potencialmente, el bloqueo de las acciones de LTB-4, mediante antagonistas de sus receptores o inhibidores de su síntesis, podría tener un papel terapéutico importante en la aterosclerosis.
Por otro lado, la ciclooxigenasa 2 (COX-2) es la enzima encargada de la conversión del ácido araquidónico en prostanoides. PGG2 es la primera prostaglandina de la cascada y puede convertirse en otras, como PGI2, PGE2 y tromboxano A2 (TXA2), que tienen efectos opuestos en la biología de la pared vascular (fig. 4). PGI2 es vasodilatador y antiaterogénico, mientras que PGE2 tiene propiedades proinflamatorias y quimioatractantes20,21. PGI2, junto a NO, previene episodios trombóticos y el remodelado crónico de la pared. Por otra parte, TXA2 es protrombótico y vasoconstrictor. En pacientes con aterosclerosis o DM2, se produce la supresión de la liberación de PGI2 y, además, se liberan prostaglandinas deletéreas, como TXA2, el inhibidor del activador del plasminógeno 1 (PAI-1) y aniones superóxido21,22. PGI2 y PGE2 se sintetizan por las células endoteliales, mientras que TXA2 se produce por las plaquetas. Debido a que el precursor para PGI2 y PGE2 es el mismo, actualmente se está estudiando la posibilidad de bloquear específicamente las acciones de PGE2 a través de la inhibición de su enzima PGE2 sintasa (PTGES) o del bloqueo de sus receptores EP1 y EP3. El equilibrio de la expresión de todos estos mediadores será clave en el desarrollo de la aterosclerosis18,21.
Además, la expresión de COX-2 la regula, entre otros factores, NF-κB, y le afecta la actividad del NO y NO sintasa 23. Citocinas de la familia de TNF inducen la expresión de COX-2 y la liberación de PGE2 de modo dependiente de la dosis y el tiempo a través de la activación de NF-κB. El tratamiento con un antioxidante (bloquea activación de NF-κB) atenúa la expresión de COX-2 y la liberación de PGE224.
Modulación terapéutica de COX-2 y 5-LOX en la aterosclerosisCOX-2 está frecuentemente expresada en diversas enfermedades inflamatorias, como aterosclerosis18. El tratamiento con estatinas es capaz de disminuir la expresión de COX-2 de células circulantes y placas ateroscleróticas en modelos experimentales25 y en humanos26. Además, estos fármacos reducen los valores plasmáticos de PGE2 en pacientes con aterosclerosis carotídea26 o tras presentar un episodio de SCASEST27.
El bloqueo de la COX-2 podría ser teóricamente beneficioso en la aterosclerosis, dado que es una enfermedad inflamatoria. Sin embargo, en varios estudios clínicos, el tratamiento con inhibidores selectivos de COX-2, rofecoxib y celecoxib, aumentó el riesgo cardiovascular de pacientes con artritis reumatoide28,29, aunque con este último los resultados fueron menos homogéneos30. Este hecho podría explicarse al menos en parte debido a que TXA2 es además producido en plaquetas vía ciclooxigenasa 1 (COX-1), una enzima que era menos afectada por los inhibidores de la COX-2. En los últimos años, se han desarrollado inhibidores duales de la vía COX y 5-LOX, con el fin de evitar el componente inflamatorio y el riesgo cardiovascular asociado a los inhibidores selectivos de la COX-2. Estos fármacos, entre los que está licofelone, inhiben de forma específica la COX-2, 5-LOX y además la COX-1, y de este modo interfieren con la entrada de los leucocitos en la pared celular. En un modelo de aterosclerosis en conejo, hemos observado que el tratamiento con licofelone era más efectivo que rofecoxib con la reducción de la relación íntima/ media y la inflamación en la placa de ateroma18. Estos inhibidores duales también disminuyen la expresión de las proteínas quimioatractantes, como MCP-1. Por tanto, es posible que el efecto beneficioso de estos fármacos se deba al bloqueo dual de la formación de prostanoides, como PGE2, TXA2 y LTB-4, por lo que es posible que puedan evitar el riesgo cardiovascular de inhibidores selectivos para COX-2.
Papel de las proteínas chaperonas HspLas proteínas HSP son una familia de 20–25 moléculas expresadas por las células en respuesta al estrés provocado por diferentes agentes, como elevada temperatura, ROS, toxinas y LDL oxidadas. Las HSP funcionan además como proteínas chaperonas (protectoras), ya que están implicadas en la renaturalización de proteínas dañadas o que han perdido su conformación. Además, estas moléculas están involucradas en la respuesta inmunitaria y, por tanto, pueden acelerar la aterogenia31.
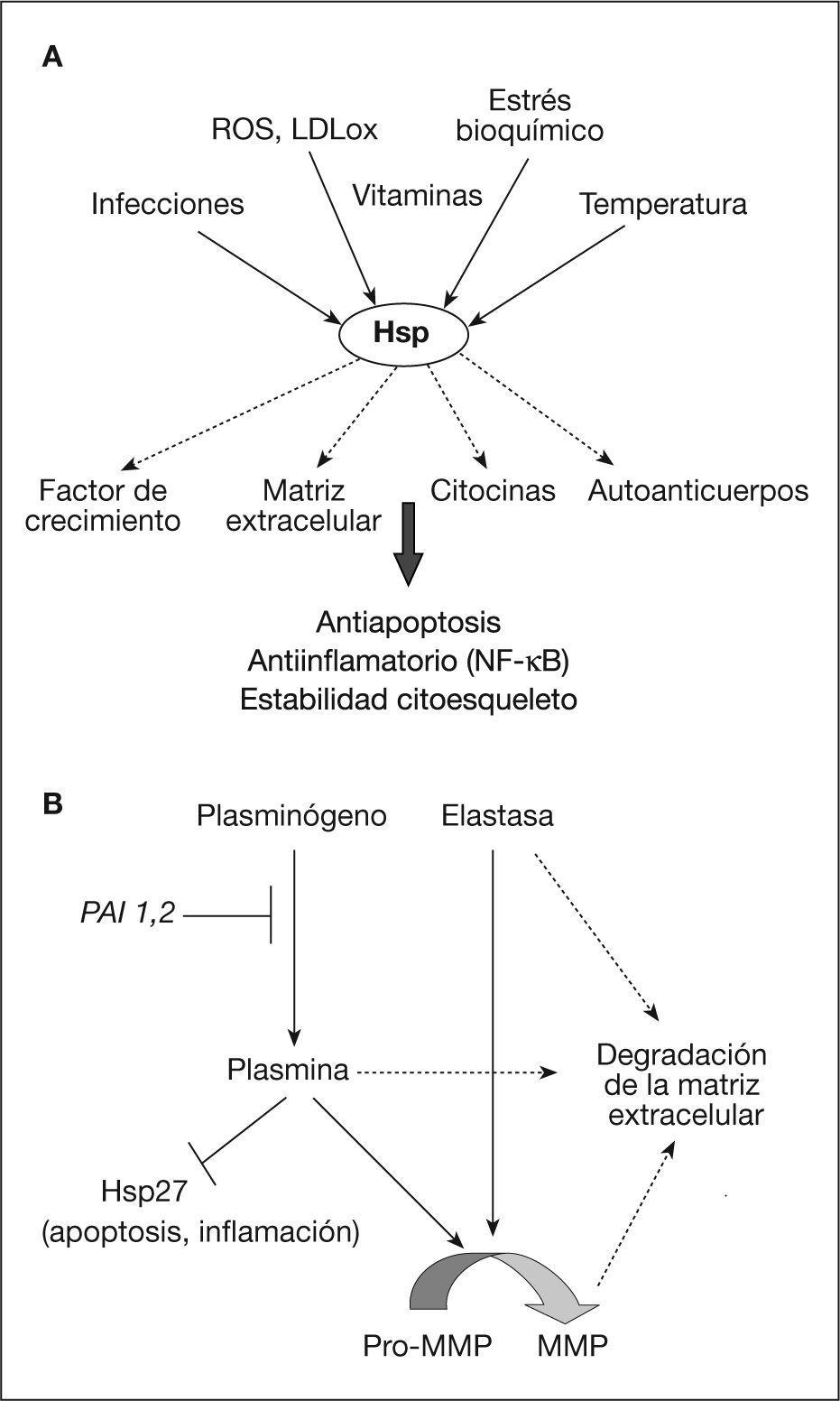
En la superficie de los macrófagos se expresan los receptores llamados toll-like receptors (TLR), que están implicados en la iniciación de la respuesta inmunitaria. En la aterosclerosis, se ha observado expresión de TLR1-5, que sirven como receptores para lípidos modificados y HSP32. En situaciones de descompensación nutricional, como en dieta rica en grasa o anormalidades del metabolismo lipídico, donde hay un incremento de los valores circulantes de LDL y/o colesterol, se induce la expresión de las Hsp33. Además, las Hsp son capaces de modular la apoptosis celular (fig. 5A). En este sentido, se conocen varias vías apoptóticas (Daxx-Fas, citocromo C-DIABLO) que se bloquean por Hsp27. La apoptosis de las CML es un episodio clave en la desestabilización de la placa, ya que desempeña un papel importante en la formación de core necrótico. El papel antiapoptótico de las Hsp puede también deberse a su capacidad de modulación de la actividad del citoesqueleto34. Hsp27 es una proteína pequeña y oligomérica que, tras ser fosforilada, se asocia a la actina citosquelética y estabiliza sus filamentos en situaciones de estrés (protección).
. Diferentes estímulos desencadenan la síntesis y la activación de proteínas Hsp. Éstas responden protegiendo de la inflamación, la muerte y la desorganización celular. B. Regulación de la degradación de la matriz extracelular. La síntesis de proteasas y metaloproteinasas (MMP) produce la degradación de proteínas de matriz extracelular de la capa fibrosa (desestabilizando la placa). La plasmina, además, proteoliza a Hsp27, inhibiendo su función protectora, antiinflamatoria y antiapoptótica. LDLox: lipoproteínas de baja densidad oxidadas; NFκB: factor nuclear de transcripción kappa B; ROS: radicales de oxígeno.")
A. Activación de las Hsp (del inglés heat shock proteins). Diferentes estímulos desencadenan la síntesis y la activación de proteínas Hsp. Éstas responden protegiendo de la inflamación, la muerte y la desorganización celular. B. Regulación de la degradación de la matriz extracelular. La síntesis de proteasas y metaloproteinasas (MMP) produce la degradación de proteínas de matriz extracelular de la capa fibrosa (desestabilizando la placa). La plasmina, además, proteoliza a Hsp27, inhibiendo su función protectora, antiinflamatoria y antiapoptótica. LDLox: lipoproteínas de baja densidad oxidadas; NFκB: factor nuclear de transcripción kappa B; ROS: radicales de oxígeno.
Hay evidencias de la inhibición de NF-κB por proteínas Hsp35. En macrófagos, se detectaron Hsp tras media hora de aumento moderado de temperatura y permanecieron así hasta 24 h tras el daño, en correlación con una disminución de la actividad de NF-κB36. Es posible que las acciones de las Hsp atenúen la activación de la respuesta inflamatoria aguda de NF-κB. La inhibición de la activación de NF-κB mediante Hsp dependerá del grado y la duración del daño. Sin embargo, Hsp27 puede ser escindida por la plasmina, que se libera junto a otras proteasas (metaloproteinasas) durante la progresión de la lesión aterosclerótica. Estas proteasas pueden liberarse ya en forma activa desde las células inflamatorias, como es el caso de la elastasa, o generadas a partir de formas precursoras circulantes, como en el caso de la plasmina (fig. 5B). La acción de las proteasas, además de inducir desestabilización en la placa (al degradar la capa fibrosa), limita el papel protector del la Hsp2734. Mediante el análisis proteómico del sobrenadante del cultivo de placas obtenidas por endarterectomía de pacientes, hemos observado la disminución de los valores de Hsp27 comparado con fragmentos de arteria sana. Al restaurar Hsp27 con proteína recombinante, se confirmó que ésta era degradada posiblemente por las proteasas liberadas de la placa37. Además, en el plasma de pacientes con aterosclerosis, Hsp27 se encuentra en valores inferiores respecto a individuos control38.
Conclusiones y perspectivasLa enfermedad aterotrombótica se caracteriza principalmente por un componente inflamatorio, que es la causa de su evolución. A partir de este episodio inflamatorio, en los últimos años se están investigando nuevos mediadores, como los miembros proinflamatorios de la familia del TNF (TWEAK), enzimas encargadas de la síntesis de prostanoides (5-LOX y COX-2) y agentes protectores de apoptosis y desnaturalización proteica (Hsp). Todos ellos participan en las diferentes fases del desarrollo de la lesión aterosclerótica y es posible que su modulación pueda impedir la formación de la placa o su desestabilización y, por consiguiente, la formación del trombo. Un tratamiento farmacológico específico y selectivo frente a estas dianas podría suponer un gran avance en la prevención y el tratamiento de la aterosclerosis.
Conflicto de intereses: Los trabajos de los autores citados en el texto se han financiado con fondos de la Fundación Española del Corazón, Fondo de Investigaciones Sanitarias (PI050451 y CP04/00060), Fundación Ramón Areces, Sociedad Española de Arteriosclerosis, Ministerio de Educación y Ciencia (RyC 2005-896, SAF2007/63648 y SAF 2007/60896), CAM (S2006/GEN-0247) y Pfizer.