




Como enfermedad cardiovascular prevalente, la insuficiencia cardíaca es una de las principales causas de morbimortalidad prematura. Por ello, existe un especial interés sobre el estudio de marcadores eficientes asociados al riesgo y/o predicción de eventos cardiovasculares. En consecuencia se proponen a múltiples candidatos, pero sobresalen especialmente aquellos implicados en procesos oxidativos e inflamatorios propios de la enfermedad cardiovascular como el anión superóxido, óxido nítrico y peroxinitrito. En este sentido, existe una falta de conocimiento sobre las potenciales utilidades de estos sistemas como biomarcadores. La presente revisión procura contribuir a la mayor comprensión de estos sistemas para una mejor caracterización de pacientes. Por otra parte, un profundo conocimiento de estos complejos sistemas también permitiría proponer nuevas líneas de investigación para el desarrollo de inéditas herramientas terapéuticas como una auspiciosa frontera para el abordaje de esta patología.
As a prevalent cardiovascular disease, heart failure is one of the leading causes of morbidity and premature mortality. Therefore, there is a special interest in the study of efficient markers associated with risk and / or prediction of cardiovascular events. Multiple candidates are proposed, especially those involved in oxidative and inflammatory processes typical of cardiovascular disease, such as superoxide anion, nitric oxide, and peroxynitrite. There is a lack of knowledge on the potential usefulness of these systems as biomarkers. This review aims to contribute to a better understanding of these systems, as well as an improved patient profile. Furthermore, a deep knowledge of these complex systems would also allow proposing new lines of research for the development of new therapeutic tools as a promising start for new approaches to this disease.
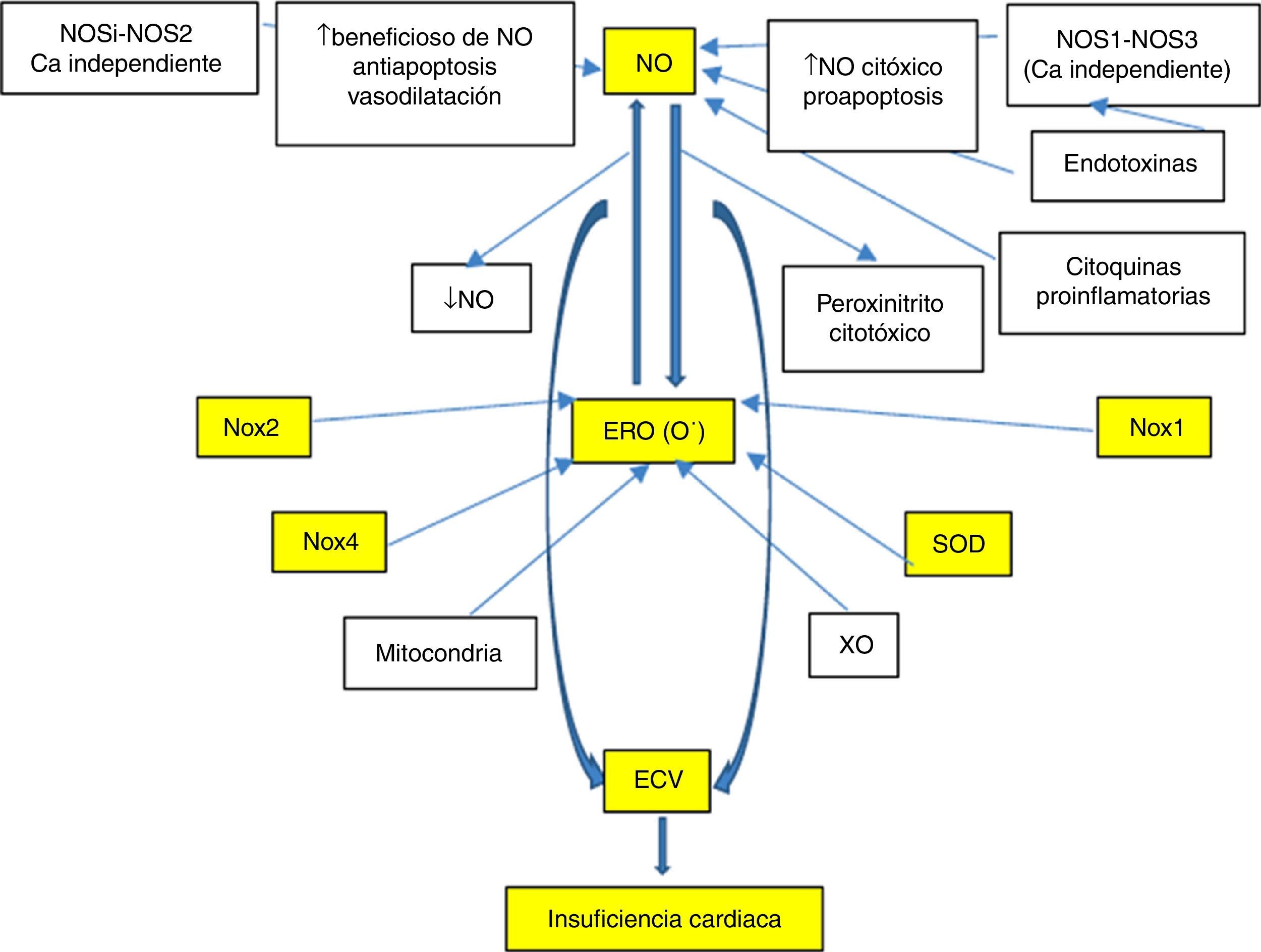
En relación a la enfermedad cardiovascular (ECV), existe un especial interés sobre el estudio y desarrollo de marcadores eficientes asociados al riesgo y/o predicción de eventos, que a su vez posibiliten una oportuna intervención, lo que finalmente valide su potencial de predicción de riesgo cardiovascular. Este contexto conduce la búsqueda de nuevos biomarcadores en donde se discuten y proponen a múltiples candidatos; no obstante sobresalen especialmente los implicados en procesos oxidativos e inflamatorios inherentes a la ECV. De hecho, las especies reactivas de oxígeno (ERO), en especial el anión superóxido (O−) y las especies reactivas de nitrógeno (ERN), tales como el óxido nítrico (NO) y peroxinitrito, manifiestan funciones protagónicas (fig. 1). En este sentido, y con especial énfasis sobre el conocimiento de las vías de señalización implicadas en la fisiopatología de la insuficiencia cardíaca (IC), existe una falta de conocimiento sobre las potenciales utilidades de estos sistemas como biomarcadores. La mayor comprensión posibilitaría una mejor caracterización y también abriría nuevas líneas de investigación para el desarrollo de inéditas herramientas terapéuticas, lo que podría significar una auspiciosa frontera para el abordaje de esta patología.

Específicamente se han investigado distintos sistemas neurohumorales para identificar biomarcadores con buena capacidad predictora, sin embargo, pocos cumplen con todos los criterios requeridos para que aporten utilidad a la clínica. Surge entonces la necesidad de continuar en la búsqueda de más y mejores sustancias que constituyan una contribución superadora.
En los últimos años, ha sido reconocida a la ECV como un «continuo» que involucra una multiplicidad de entidades tales como la enfermedad primaria del músculo cardíaco (miocardiopatía), hipertensión arterial (HTA), hipertrofia ventricular izquierda (HVI), enfermedad coronaria ateroesclerótica, arritmias cardíacas, diabetes mellitus; y donde la IC resulta la vía final común de todas ellas con alteración de las vías de señalización que involucran al NO, ERO/ERN, NADPH oxidasa (Nox) y superóxido dismutasa (SOD).
La IC es frecuentemente abordada desde la perspectiva de los principales mecanismos que inducen daño y remodelado ventricular como resultado de la sobreestimulación neurohumoral a partir de los sistemas renina-angiotensina-aldosterona (RAS) y adrenérgico. Estas alteraciones resultan clásicas y marcan la evolución de la enfermedad. De particular interés para la presente revisión, el aumento de la resistencia vascular periférica y el remodelado cardiaco constituyen las principales alteraciones y se encuentran condicionadas a las vías de señalización que previamente fueran mencionadas. Por lo tanto, resulta novedosa la propuesta de evaluar la evolución de la IC pero desde una perspectiva inédita para la práctica clínica como lo es el formado por las vías de señalización que implican al ON, O-Nox y SOD. En acuerdo con esta sugerencia, ha sido recientemente referido que el deterioro en las vías de señalización que involucran al NO y factores relacionados, estarían asociados a pronóstico y/o mortalidad durante la IC.
Por otro lado, el uso de biomarcadores clásicos en IC y las drogas para su tratamiento, resultan costosos y de difícil acceso para el paciente principalmente en el ámbito de la salud pública. Consecuentemente, la IC resulta una enfermedad de importancia epidemiológica, sanitaria y económica. Esto justifica el apoyo para la realización de grandes esfuerzos de investigación a los efectos de su mejor comprensión, tratamiento y seguimiento. Por lo que el desarrollo de nuevas metodologías de estudio sobre evolución/pronóstico, más accesibles y de menor costo, permitirían alterar positivamente la evolución natural de este síndrome. La implementación de biomarcadores de esta naturaleza –utilizados para mejorar la expectativa y calidad de vida– podrían además colaborar con estudios sobre evaluación de drogas y dispositivos electrofisiológicos durante la IC.
Biomarcadores en insuficiencia cardíacaEn términos generales los biomarcadores proporcionan información pronóstica útil en pacientes con IC y actualmente revisten un interés considerable en la determinación de su capacidad para guiar la terapia en casos de IC aguda y crónica. Bajo estos preceptos, Richards y Braunwald enumeraron marcadores neurohumorales, inflamatorios, de estrés oxidativo, de remodelado de matriz intersticial, de lesión miocitaria y otros considerados más novedosos, que reflejan distintos aspectos fisiopatológicos de la IC1,2.
Actualmente son de preferencia por su valor diagnóstico/predictivo en IC, los marcadores neurohumorales conocidos como péptidos natriuréticos cardíacos de tipo B (BNP, NT-proBNP y proBNP) que se producen en el ventrículo izquierdo (VI) y en el ventrículo derecho (VD), y que específicamente informan sobre presión de llenado del VI y tensión parietal. Sin embargo, también aumentan cuando el paciente tiene falla derecha sin falla de VI como por ejemplo se observa en las enfermedades respiratorias con cor pulmonar crónico y en la hipertensión arterial pulmonar del grupo 1 (Niza). Estos marcadores permiten excluir IC reduciendo imprecisiones y costos, siendo su determinación seriada una herramienta útil para identificar el momento adecuado para el alta y la evolución postalta.
Por lo expuesto, los biomarcadores en general resultan útiles pero también muestran limitaciones al momento de su utilización. Su utilidad clínica está relacionada con que la determinación facilite el manejo clínico y mejore el pronóstico de una o más de situaciones como para mejorar la certeza diagnóstica, asociación al riesgo de aparición o agravamiento de IC –aquí lo ideal es que conlleve una respuesta con un tratamiento específico–, también la monitorización a través de determinaciones seriadas de marcadores debería mejorar los resultados obtenidos en el seguimiento del paciente, es decir menos descompensaciones agudas, reducción de la mortalidad y/o mejora de la calidad de vida.
Por otro lado, el uso de biomarcadores en IC plantea el rol del laboratorio y el valor de una simple muestra de sangre en el diagnóstico, pronóstico, monitoreo de la evolución y para guiar la terapia. De este modo, el mayor conocimiento obtenido sobre el protagonismo de los sistemas neurohormonales en la evolución de la IC, ha permitido lograr importantes progresos terapéuticos desde mediados de los ochenta. Esto se ha basado en el estudio de las concentraciones circulantes de marcadores de tensión miocitaria, BNP-proBNP y NT-pro BNP, que resultaron ser además, los más cercanos a los clásicos postulados del biomarcador ideal. Sin embargo, además de los marcadores mencionados, en los últimos años se investigan con especial atención a moduladores del estrés oxidativo y donde las vías de señalización del NO, Nox-ERO y la SOD manifiestan una importante participación en el desarrollo de la ECV (fig. 1). Así, un profundo conocimiento de ellos permitiría comprender más en detalle los mecanismos, cambios neurohumorales y daños biomoleculares que a la fecha no resultan completamente establecidos y que ocurren durante la IC.
Óxido nítrico como biomarcador en insuficiencia cardíacaMúltiples estudios han demostrado la participación del NO en la fisiopatología de la IC y en donde existe un marcado desequilibrio NO/redox a expensas del incremento de las vías enzimáticas que producen radicales libres como son las Nox vasculares, XO cardíaca, enzimas mitocondriales, hemoglobina oxidasa en eritrocitos, entre las más destacadas. En términos generales esto produce oxidación de las proteínas que participan del acoplamiento excitación-contracción, y también promueven una menor biodisponibilidad del NO a expensas de alterar la actividad y/o localización de las enzimas productoras (NOS y XO)3,4. Lo cual conlleva al desacople mecanoenergético característico debido a que la reducción de la contracción no se acompaña de una reducción proporcional en el consumo de energía. También existe una marcada activación neurohumoral con aumento de citocinas proinflamatorias que inducen la expresión de NOS25 y, en ocasiones, aumentan las actividades de NOS1 y NOS3 modificándose la localización de la NOS16,7. Sin embargo, la hipótesis de que el NO desempeña un importante papel en la etiopatogenia de la IC no concuerda con el hallazgo de que el mismo elemento ejerce un efecto cardioprotector frente a la isquemia o que en el miocardio insuficiente hay una menor expresión de NOS y/o menor biodisponibilidad en pacientes con IC. De hecho, en modelos experimentales y en pacientes con IC, la actividad de NOS3 puede aumentar, disminuir o mantenerse invariable4,8,9 y en donde la NOS2 aumenta marcadamente para algunos estudios, pero no en todos los casos5–7,10–14. Ello sugeriría –al menos en parte– que los cambios en la expresión de NO2 y NOS3 podrían responder a un epifenómeno que acompaña a la IC, pero sin ser determinantes de su causa (fig. 1).
En el miocardio insuficiente, la disminución del NO contribuiría a una menor vasodilatación coronaria endotelio-dependiente, a la reducción de la relajación ventricular y al aumento del consumo de oxígeno máximo (MVO2)15,16. Por lo tanto, la sobrexpresión de NOS3 debería mejorar estas alteraciones como ha sido demostrado en algunos estudios experimentales, además, tras realizar una ligadura coronaria existe una mejoría de la función contráctil y una reducción de la hipertrofia del VI y la mortalidad17–19; mientras que ratones noquedos para NOS3 en los que también se realizó ligadura coronaria, presentaron hipertrofia y dilatación del VI, disminución del acortamiento fraccional, de la fracción de eyección, con aumento del volumen telediastólico, del diámetro interno del VI, rarefacción vascular y un aumento de la mortalidad8,20. También, en estos ratones disminuyen los efectos beneficiosos de enalapril o valsartán21. Estos resultados sugieren que la NOS3 reduce la disfunción ventricular y el remodelado postinfarto, y además participaría de alguna forma en la disfunción ventricular producida por la angiotensina II.
Por otro lado ha sido comunicado en pacientes con miocardiopatía dilatada (MCD) que la disminución del NO no altera la contracción cardíaca, y donde la infusión intracoronaria de nitroprusiato de sodio o sustancia P, no modifica los parámetros hemodinámicos10,22–24. Sin embargo, en un modelo animal de IC inducido por sobrestimulación se pudo demostrar que la fase de descompensación cardíaca se caracterizaba por una marcada reducción de la producción de NO, de la dP/dt máx y de la distensibilidad ventricular (que aumenta la presión telediastólica del VI con aumento del MVO2), a la vez que el metabolismo cardíaco pasa de utilizar ácidos grasos a utilizar glucosa como fuente de energía15; estos resultados sugieren que el NO participaría del acoplamiento entre el flujo coronario, la actividad contráctil y el metabolismo cardíaco.
En pacientes con MCD aumenta la expresión de NOS1, que se transloca desde el retículo sarcoplámico (donde se encuentra acoplada a XO) al sarcolema, donde al igual que sucede con la NOS3, se asocia con una ATP-asa calcio dependiente regulada por calmodulina7. Estos cambios de expresión y localización pueden ser beneficiosos durante la IC, ya que NOS1: a) inhibe las respuestas cardíacas a la estimulación de los receptores β y al inhibir la corriente de Ca++, y quizás la liberación de Ca++ desde el retículo sarcoplásmico, podría ejercer un efecto protector frente a la cardiotoxicidad de las catecolaminas25. b) Aumenta el tono vagal cardíaco, disminuyendo la frecuencia cardíaca26. c) Restaura la actividad baroceptora central; y d) podría compensar la inhibición de NOS37. Sin embargo, es posible también que el desplazamiento de NOS1 al sarcolema facilite el estrés oxidativo, ya que se perdería el control que ejerce sobre la actividad de XO en el retículo sarcoplásmico y además esto alteraría el balance óxido/redox del sarcolema9.
De interés, durante la IC se observa una reducción de la respuesta inotrópica a la estimulación β como consecuencia de alteraciones en la densidad de receptores (disminuyen los receptores β1 y β2, y aumentan los β3 que median respuestas inotrópicas negativas) o en el acoplamiento del receptor con sus vías de señalización (aumenta la expresión de la cinasa específica del receptor β-adrenérgico o ARK y de proteínas Gi, y disminuyen las Gs). Los β3 son más resistentes a la desensibilización homóloga que representa el aumento del tono simpático característico de la IC, por lo que su estimulación facilitaría la producción continua de NO en presencia del incremento del tono simpático existente en la IC27. Por tanto, el NO sintetizado a través de la vías β3-NOS3, NOS1 translocado al sarcolema, y de la inducción de la NOS2, modularían la respuesta a las catecolaminas y antagonizarían su toxicidad en el miocardio insuficiente. De hecho, la inhibición de las NOS potencia el aumento contráctil producido por los agonistas β-adrenérgicos en modelos animales16 y en pacientes con IC9,28,29. Sin embargo, resulta lógico suponer que el NO es tan solo uno de los factores que regulan la respuesta a los agonistas β-adrenérgicos en el paciente con IC30. No obstante, en determinadas circunstancias el aumento de NO sintetizado tras la inducción de la NOS2 puede ser beneficioso ya que mejora la relajación ventricular6,22 reduce el MVO231, la respuesta a la estimulación β-adrenérgica8,11,15 y aumenta la angiogénesis32. En cardiomiocitos de pacientes con trasplante cardíaco, el isoproterenol produce un discreto aumento de la contractilidad y frecuencia cardíaca, y donde la inhibición de NOS2 normaliza ambas respuestas, así como el tránsito de Ca++. Sin embargo, la inhibición de NOS2 no produce ningún efecto en cardiomiocitos normales o en cardiomiocitos de pacientes con IC, en los que la respuesta al isoproterenol estaba conservada y en los que la expresión de NOS2 es pobre14. Es decir, en pacientes con IC la expresión de NOS2 limita la respuesta a los agonistas β-adrenérgicos, un efecto que podría estar mediado por la inhibición de la liberación de Ca++ desde el retículo sarcoplásmico a través de una vía independiente del GMPc, pero relacionada con modificaciones del estado redox celular producidas por el peroxinitrito33. En ratones en los que se sobreexpresa NOS2 cardíaca aumenta la generación de peroxinitrito y se observa dilatación, hipertrofia y fibrosis cardíaca; más aún, aunque raramente desarrollan IC, sí presentan una alta incidencia de muerte súbita asociada con bradiarritmias34. Por otro lado, ratones con sobreexpresión de NOS2 presentan un genotipo normal, ya que el NO producido es neutralizado por la mioglobina citoplasmática; mientras que por el contrario, cuando se repiten estos experimentos en ratones carentes de mioglobina, los animales presentan hipertrofia, fibrosis intersticial y dilatación ventricular35.
Como fuera mencionado, durante la IC aumentan las vías enzimáticas que producen radicales libres y se alteran también las enzimas productoras de NO (NOSs y XO), lo que produce vasoconstricción y desacople mecanoenergético. De hecho, en modelos animales, la transición hacia la IC descompensada implica un déficit de NOS1 y NOS3 –y por ende– en la síntesis cardíaca de NO, más un aumento en la actividad de la XO4. El NO también regula una Nox que inhibe la liberación de Ca++ por el retículo sarcoplásmico, por lo que el balance entre NO y estrés oxidativo también regula la función cardíaca a través de sus efectos sobre la señalización intracelular del catión Ca++. La XO, una importante fuente de radical superóxido, aumenta en el miocardio y los vasos de los pacientes con IC36, produciendo disfunción endotelial, depresión de la función cardíaca, desacople mecánico-energético y apoptosis37,38. La inhibición de XO con allopurinol mejora la eficiencia mecánica, el remodelado postinfarto de miocardio y la respuesta a las catecolaminas39. Aquí se destaca que los efectos del allopurinol se suprimen al bloquear NOS con L-NAME (inhibidor específico) y los de esta con alopurinol38, lo que indica que existiría interacción entre ambas vías de señalización4. Por otro lado, algunos fármacos como las estatinas40 y los IECA41 utilizados en IC, modifican el balance NO/redox al aumentar los valores de bradicinina y la síntesis de NO, y reducir la producción de radicales libres (superóxido, peroxinitrito) al inhibir la NADPH oxidasa (fig. 1).
Por otro lado, el NO tiene también un importante rol sobre los canales iónicos cardíacos, en la génesis de las arritmias cardíacas, en la apoptosis cardíaca, el precondicionamiento isquémico, la isquemia cardíaca y la función mitocondrial. En donde el daño o beneficio que NO modularía, dependerá del tipo y condición celular. Así, altas concentraciones inducen muerte celular durante la injuria isquémica y como consecuencia de enfermedades neurodegenerativas42–44.
La citoxidad atribuida al NO involucra la generación de peroxinitrito producida por la reacción de difusión controlada entre NO y el anión superóxido. El peroxinitrito interactúa con lípidos, DNA y proteínas, vía reacción oxidativa directa o indirecta por mecanismos mediados por radicales. Estas reacciones disparan respuestas celulares que van desde modulaciones sutiles de señalización celular a una abrumadora injuria oxidativa llevando a necrosis o apoptosis celular. El peroxinitrito resulta crucial en condiciones tales como stroke, infarto de miocardio, insuficiencia cardíaca crónica (ICC), diabetes, shock cardiogénico, enfermedades inflamatorias crónicas, cáncer y enfermedades neurodegenerativas45.
El efecto cardioprotector responde al menos en parte a la vía que involucra la kinasa G (GMP/proteína PKG)46,47. Donde NO activa una guanilato ciclasa soluble que cataliza la síntesis de cGMP a partir de GTP. Además, NO participa en mecanismos de acciones antiapotóticas mediadas por PKG, y estos son un área activa de investigación y donde la modulación de dichas vías tendrían importantes implicancias terapéuticas.
Factores relacionados a óxido nítrico como biomarcadores en la insuficiencia cardíacaEn pacientes con falla cardíaca crónica, ha sido claramente demostrado el incremento en la producción de ERO y donde se destaca al O−. La evidencia demuestra el papel de las ERO, junto con las ERN en la fisiopatología de la ECV. Por lo que se evalúan múltiples biomarcadores de estrés oxidativo que resultan comunes a ambos sistemas. De ellos, la Nox representa al sistema más eficiente y mayor fuente productora de ERO durante la IC. La activación anormal del RAS con impacto en la inducción de la apoptosis y fibrosis, resulta como consecuencia de la generación de ERO por Nox y es propio de las EVC48,49. Al respecto, se ha enfatizado sobre alteraciones de la vías de señalización ERO-Nox-dependiente, como un importante factor responsable del desarrollo de muchos procesos patológicos cardíacos. Así, las Nox son la mayor fuente productora de O− en células vasculares y miocitos, donde comparten algunas de las características de las enzimas presentes en los neutrófilos. En respuesta a factores de crecimiento y citoquinas, producen O− que puede ser metabolizado a peróxido de hidrógeno. Estas dos especies reactivas del oxígeno sirven como segundos mensajeros para activar múltiples vías de señalización intracelular. Además de la Nox de los miocitos, también existe una Nox vascular que ha sido reconocida esencial en la respuesta fisiológica de las células vasculares, incluyendo el crecimiento, migración y modificación de la matriz extracelular. Además, también se la ha relacionado con HTA y estados patológicos asociados a crecimiento descontrolado e inflamación, tal como la ateroesclerosis. En el caso de sobrecarga de presión se activa la producción de Ang II miocárdica, la cual estimula vías de señalización intracelular que activan la respuesta hipertrófica, tal y como lo realiza Nox50.
Nox tiene diversos constituyentes o subunidades que resultan relevantes por su relación con la ECV, siendo en orden de importancia la Nox2, Nox4 y Nox1 y las subunidades gp91phox, p22phox, p47phox y Rac1. Estas desempeñan un importante rol en la injuria cardíaca, participando en la producción de hipertrofia cardíaca, produciendo respuestas celulares opuestas, acelerando el proceso ateroesclerótico, HTA y la remodelación miocárdica, activándose en IC y post-IAM50–52.
Más específicamente, Esposito et al.53 evaluaron ratones a los cuales se les había eliminado Rac1 y llegaron a la conclusión de que esto previno la hipertrofia inducida por Ang II. Rac1 iniciaba la respuesta hipertrófica dependiente de ERO generada por Nox en corazón54, confirmándose que la producción de O− por la subunidada Rac1 de Nox2, inició la activación de protein kinasa B (Akt) como componente de esta vía de señalización e inducida por Ang II para la hipertrofia de los cardiomiocitos55.
La investigación básica provee evidencias sobre Nox2 como responsable de la producción vascular de ERO, de una menor biodisponibilidad de NO y del desarrollo de las lesiones precoces en las aortas de ratones56. Un hallazgo adicional agrega la participación del factor transformador de crecimiento beta (TGF-β) circulante y de la apolipoproteína E, y en donde el aumento de TGF-β indujo la activación de Nox y sobreproducción de ERO acelerando el proceso ateroesclerótico, la HTA y la remodelación miocárdica en ratones deficientes en apolipoproteína E57.
Por otro lado, una elevada expresión de Nox y O− fueron también descriptas en carótidas de conejos con ICC58 y específicamente en la actividad de la subunidad p47phox de Nox2 en VI de ratones después del infarto de miocardio (IM)59.
Mientras que Nox2 está involucrada en la HVI inducida por angiotensina II, Nox4, también lo estuvo aparentemente en la sobrecarga de presión en miocardio murino60. Esta isoforma entonces aunque con controversias, también desempeñaría un importante rol en la ECV. Fue sugerido que Nox4 estaba unida a la proteína p22phox sobre la membrana interna en las células epiteliales. También se halló –en contraste con otras isoformas de NADPH oxidasa– que Nox4 producía principalmente peróxido de hidrógeno y una muy pequeña cantidad de O−. Proteínas citosólicas oxidasas o GTPasa Rac no fueron requeridas para la actividad de esta enzima61. En este sentido, existe alguna evidencia mencionando que si bien Nox4 produjo peróxido de hidrógeno, también generó O− intracelularmente62. Pero sigue existiendo mucha incertidumbre sobre la producción de ERO-Nox4-dependiente. Por ello, el estado actual del conocimiento a este respecto refiere que Nox4 produce principalmente peróxido de hidrógeno y no O- contradiciendo a la mayoría de otros datos experimentales. Una explicación factible sería el uso de métodos no fiables tales como la reducción de nitroblue tetrazolium para la detección de O−61–63. Por otro lado, el empleo de métodos específicos y precisos como la quimioluminiscencia para detección de O- arrojaron diferentes resultados64. Por ejemplo, con lucigenina quimioluminicente se encontró que Nox4 y Nox2 producían aproximadamente el 75% de O− en las arterias coronarias de pacientes con enfermedad arterial coronaria65.
Nox4 está localizada en diferentes sitios –dentro de las células cardíacas– en comparación con otras Nox; en especial se la ubica en mitocondria y donde representa una fuente mayoritaria de producción de O− en miocitos. Puede provocar disfunción mitocondrial, apoptosis, disfunción ventricular izquierda en respuesta a sobrecarga de presión y paradójicamente también produciría adaptación cardíaca al estrés crónico66. En ratones añosos bajo estimulación hipertrófica con sobrecarga de presión, Nox4 fue estimulada y esto incrementó la producción de O− e indujo disfunción cardíaca con fibrosis y apoptosis67.
En contraste con otras isoformas de NADPH oxidasa, la estimulación de Nox4 en cardiomiocitos condicionó protección contra el remodelado cardíaco inducido por sobrecarga de presión. Los autores justifican estos resultados por la preservación de la densidad capilar del miocardio inducida por Nox4 a través de la activación del factor inducible 1 (Hif1) de hipoxia y de la producción de factor de crecimiento del endotelio vascular. Ellos también mostraron que la localización de Nox4 en los cardiomiocitos, no fue mitocondrial, sino en el retículo endoplásmico perinuclear. Estos hallazgos contradicen los de otros autores66. Apareciendo resultados contradictorios concernientes a los efectos sobre el estrés oxidativo y sobre su localización. El origen de estas diferencias requiere mayor investigación68 porque se oponen a la mayoría de los hallazgos sobre los efectos de Nox en cardiomiocitos y otras células.
En contraste con las implicancias de Nox en las enfermedades cardíacas, la participación de XO en la injuria cardíaca ERO-dependiente causó originalmente muchas dudas. La XO y la xantino deshidrogenasa (XDH) son las formas oxidada y reducida de xantino-oxidoreductasa (XOR). XO fue considerada como la mayor fuente productora de O− y peróxido de hidrógeno; su mecanismo fue inicialmente bien establecido63. Paradójicamente, estudios subsecuentes descubrieron una pobre actividad de XO en corazones de animales y humanos69,70.
Sin embargo, en la actualidad existe acuerdo sobre aumento en los niveles de XO y su actividad en el sistema cardiovascular bajo condiciones patológicas, pero que no pueden ser fácilmente detectados en condiciones fisiológicas. Así Thompson-Gorman y Zweier midieron la generación de ERO mediado por XO en corazón aislado de rata71. Ellos hallaron que XO fue un importante factor de daño oxidativo por isquemia/reperfusión en corazón de rata. En este sentido, se ha demostrado también que el aumento en la producción de ERO catalizada por XO durante la isquemia/reperfusión resulta del incremento de la concentración del sustrato (xantina e hipoxantina) debido a la degradación de ATP durante la isquemia72. Ashraf y Samra también sugirireron que la actividad de XO aumenta durante la isquemia y se intensificó después de la reperfusión73. XO se localiza en células intersticiales, endotelio vascular coronario y células musculares lisas. De Jong et al.74 mostraron que la producción de ERO por xantino óxidoreductasa (XOR) aumentó en la IC pero no en la hipertrofia cardíaca.
Similarmente a las NADPH oxidasas, la XO condicionó a muchos desórdenes cardíacos ERO-dependientes como deterioro de la vasodilatación mediada por endotelio, aumento de la actividad de XO en MCD, disminución de NO en pacientes con enfermedad coronaria, disfunción endotelial coronaria y efecto tóxico de O− generado por XO en corazón. Además, el aumento de la actividad de XO y la disminución de la actividad superóxido dismutasa extracelular (ecSOD) deterioraron la vasodilatación mediada por endotelio en pacientes con IC75. Al respecto, trabajos posteriores determinaron los niveles de la proteína XO y O− dependiente de XO inducidas por Ang II en células endoteliales de pacientes con enfermedad coronaria76, sugiriéndose que la Ang II promueve sobreproducción de O− por activación de XO redox-sensible. También, estudios básicos confirmaron el rol de XO, encontrándose que su actividad estaba elevada en MCD77 y que la inhibición crónica de XO por allopurinol suprimía la progresión de IC en MCD.
También ratas con IC por HTA espontánea exhibieron aumento de la expresión de ARNm y de la actividad de XOR, mientras que la inhibición de XOR causó reversión del remodelado en IC por HTA espontánea y MCD dilatada78. La XOR y Nox también aumentan la formación O− cardíaco ratas Dahl hipertensas sal-sensibles con IC diastólica79.
Las ERO producidas por XO redujeron la biodisponibilidad de NO coronario en pacientes con EAC80. Del mismo modo, en modelo murino la isquemia/reperfusión miocárdica aumentó la expresión de factor de necrosis tumoral (TNF-α) e indujo activación de XO y generación de O− llevando a la disfunción del endotelio coronario81. Adicionalmente, se demostró también otro efecto tóxico de O− generado por XO en corazón de ratas con IC espontánemente hipertensa con MCD. Específicamente, el O− deterioró la S-nitrosilación del receptor de rianodina (RyR) siendo responsable del escape de calcio desde el retículo sarcoplásmico en músculo esquelético82.
De particular interés y amplio reconocimiento, la disfunción mitocondrial resulta una esencial fuente productora de ERO en la ECV por lo que la mitocondria permanece en constante estudio. De hecho, se estableció que el O− generado por la mitocondria resulta como consecuencia a la liberación de electrones transportadores de la cadena respiratoria (complejos I y III)83. Muchos autores confirmaron la importancia de la sobreproducción de ERO mitocondrial en el corazón dañado (miocardio insuficiente, isquemia, isquemia/reperfusión, disfunción endotelial coronaria producida por superóxido en IC congestiva, progresión de HVI a HP e IC derecha, daño mitocondrial y de VI, y por último arritmias).
Originalmente, el complejo mitocondrial I fue descripto como una fuente potencial de ERO en miocardio de perros con IC84. También, en mitocondrias de corazón aislado de ratón, la isquemia aumentó la producción de ERO85. La producción de O− mitocondrial en corazón dañado también puede inducir alteración del complejo II en miocardio postisquémico en ratas sujetas a ligadura coronaria seguida de reperfusión86. De interés, la injuria por isquemia/reperfusión con ablación de la proteína p66 (Shc) en corazón de ratón tiene un rol clave en la formación de ERO mitocondrial87. Al respecto, la despolarización mitocondrial y el aumento de la producción de ERO mediada por la lipoxigenasa y el ácido araquidónico indujo arritmias por isquemia/reperfusión88. Además, el aumento en la producción de O− mitocondrial fue responsable de disfunción endotelial coronaria y de la disminución del flujo coronario en IC congestiva89.
Para destacar, estudios realizados durante la progresión de la HVI a la IC congestiva demostraron que la Nox mitocondrial fue la principal fuente productora de ERO –especialmente en el fallo ventricular derecho inducido por HTA–; y sorprendentemente, el aumento de la actividad del complejo mitocondrial II fue el mecanismo responsable y no de los complejos I y III, reconocidos como los mayores productores de ERO mitocondrial. Este aporte fue particularmente relevante para la producción de ERO ventricular en la IC90. Sin embargo, Mariappan et al.91 demostraron que la producción de O− mitocondrial inducido por TNF-α, aumentó la actividad del complejo I de la cadena respiratoria lo que produjo daño mitocondrial en VI de ratas. Todos estos hallazgos sugieren que la sobreproducción de ERO, por la mitocondria, representan un origen causal en las enfermedades cardíacas.
Por lo expuesto, resulta claro que la ECV se asocia con un estado crónico de estrés oxidativo e inflamación mediado por vías complejas de señalización que se encuentran interconectadas. Más específicamente, la IC cursa con disfunción mitocondrial, sobreproducción de ERO, activación del RAS vinculada a mayor actividad de NADPH oxidasa y reducción del NO (fig. 1). De interés, ha sido demostrado que la menor biodisponibilidad del NO induce la expresión de proteínas de respuesta a golpe de calor tales como Hsp70, lo que condiciona efectos beneficiosos contra la lesión por estrés oxidativo, la inflamación y la apoptosis. La inducción de proteínas de golpe de calor como respuesta a daño por exaltación del sistema RAS y/o déficit de NO, fue originalmente sugerido por Bravo et al.92.
Actualmente se conoce que las proteínas de choque térmico están elevadas en plasma de pacientes con ECV, sin embargo no se comprende aún por completo su papel fisiológico y menos su valor para predecir el desarrollo y/o progresión de la enfermedad. En particular, fue sugerido que los niveles circulantes de Hsp70 podrían indicar la presencia / progresión de la aterosclerosis en sujetos con hipertensión establecida, y además una posibilidad intrigante que plantean los autores es que Hsp70 podría proteger contra el daño oxidativo e inflamatorio en este grupo de sujetos93. En este sentido, recientemente ha sido propuesto que Hsp70 sería un potencial biomarcador y objetivo terapéutico en patologías como cáncer, ECV, neurológicas y hepáticas94.
Por otro lado, fue informado que bajos niveles de Hsp70 se relacionarían con un estado cardiovascular saludable y se sugirió como predictor de longevidad95. Con estos antecedentes, Hsp70 fue investigada en pacientes con ICC intentando establecer relación entre gravedad y supervivencia. Aquí se encontró elevación en los niveles de Hsp70 particularmente en aquellos con caquexia cardíaca y por ello pudo ser relaciona con la gravedad de la enfermedad; sin embargo, no con la supervivencia96. Por lo tanto, la importancia de la relación de la expresión de Hsp70 y la morbilidad en ICC requiere mayor estudio.
Conclusiones y perspectivasLa IC es, en casi todos los casos, el resultado final de la enfermedad cardíaca primaria y cualquier causa de deterioro estructural del corazón, resultando una de las ECV más prevalentes. Dicha condición la posiciona como una de las principales causas de morbimortalidad prematura en la mayoría de los países industrializados y en vías de desarrollo. De especial interés, existe evidencia obtenida de fuentes como estudios epidemiológicos, prospectivos de cohorte e intervención, que sugieren que esta patología se asocia con alteraciones de la función endotelial, metabolismo oxidativo, inflamación y apoptosis. Entre los factores determinantes de tales alteraciones se destacan la actividad de SOD, fosforilación y expresión de enzimas productoras del NO, aumento de la actividad de la glutatión peroxidasa, activación de NADPH oxidasa y expresión de p22phox, entre los factores más relevantes.
En consistencia, la literatura destaca que pacientes con IC presentan alteración en las vías de señalización promotoras del estrés oxidativo e inflamación. Específicamente incremento de las ERO con aumento de la actividad de NADPH y déficit de la actividad de SOD. Esto fue recientemente evidenciado por nuestro laboratorio97. Por otro lado, en este mismo contexto fueron informados bajos niveles del NO. Estos podrían responder a interacción con ERO, menor producción del sistema o una suma de ambas situaciones. Si bien existe abundante bibliografía sobre alteración en las vías de señalización de NO, Nox-ERO y SOD en investigación básica, son pocos los que refieren su impacto en cuanto a su correlato en la clínica. No obstante el conocimiento actual y transcurridos más de 20 años desde la identificación de las funciones del NO endógeno, resultan infructuosos los intentos por generar nuevas estrategias terapéuticas, reflejando un lento progreso del conocimiento sobre posibles biomarcadores relacionados.
La alteración y/o desacople de Nox, ERO y SOD condicionan a múltiples vías de señalización que involucran la producción de HVI, falla miocárdica, progresión de la IC a falla cardíaca derecha, alteraciones de la función endotelial y de la vasomotilidad de las arterias coronarias e injuria de reperfusión y arritmias, todos componentes del continuo cardiovascular. En este contexto, el NO resulta clave en el mantenimiento de la homeostasis cardiovascular y por lo tanto, una alteración en su biodisponibilidad claramente se asocia con ECV. Existe evidencia sustancial –y puesta de manifiesto en esta revisión– que las ERO mayoritariamente generadas a partir de NADPH oxidasa serían responsables primarios de la reducción en la biodisponibilidad de NO. Así, el O− podrá interactuar con el ON para producir peroxinitrito que a su vez puede dar lugar a otras especies reactivas. Particularmente, el incremento del estrés oxidativo durante la IC podría resultar del desacople funcional de la cadena respiratoria como consecuencia de una capacidad antioxidante deteriorada (disminución de la actividad de SOD y/o estimulación de fuentes enzimáticas incluyendo NADPH oxidasas).
La propuesta de evaluar al NO como biomarcador en IC, planteada por nuestro grupo de trabajo y otros, respondería al supuesto de que este factor participa en aspectos relevantes fisiopatológicos de la misma. De hecho, en IC existe desequilibrio NO/redox con aumento de las vías enzimáticas que producen radicales libres (NADPH oxidasas, XO cardíaca, enzimas mitocondriales, hemoglobina oxidasa en los eritrocitos, etc.) que oxidan las proteínas que participan en el acoplamiento excitación-contracción e inactivan al NO y alteran la actividad y localización de las enzimas productoras del mismo. Ello conduce a un desacople funcional caracterizado por una reducción de la contracción que no se acompaña de una reducción similar en el consumo de energía. Todo ello, también condiciona una marcada activación neurohumoral y un aumento de citocinas proinflamatorias. En este sentido, muy recientemente ha sido sugerido que una baja biodisponibilidad del NO induce a la expresión de proteínas de respuesta a estrés oxidativo como la Hsp70 lo que promueve efectos protectores contra la lesión –no solo por estrés oxidativo–, sino también por inflamación y apoptosis. De hecho, nuestro laboratorio sugirió que Hsp70 podría utilizarse como un posible biomarcador de daño oxidativo-inflamatorio98. En consistencia, estudios recientes sugieren que las proteínas de choque térmico desempeñan un papel clave en la patogénesis de las enfermedades cardiovasculares incluyendo la IC. Al respecto, no está claro si los niveles circulantes de proteína Hsp70 están relacionados con factores de riesgo cardiovascular, índices ecocardiográficos de remodelado del VI y/o prevalencia de ECV99. Sin embargo, el análisis de resultados preliminares de nuestro laboratorio sugiere que los pacientes con IC también presentan elevación de marcadores inflamatorios (proteína C reactiva, IL-6 y TNFα), mientras que Hsp70 se encontró reducida respecto a los pacientes sanos. En consecuencia, la determinación de los marcadores oxidativos como los generales inflamatorios en plasma, nos permitieron caracterizar a pacientes sanos de los pacientes con IC. Inéditos y controversiales fueron estos hallazgos de menores niveles de Hsp70 en plasmas de pacientes con IC y que presentan bajos niveles de NO. Más compleja aún resulta la interpretación de nuestros resultados desde que se conocen las acciones opuestas que presenta Hsp70 a nivel extracelular versus intracelular, sobre la activación de la vía inflamatoria por NF-κB100. Sin embargo, con relación a estrés oxidativo e inflamación, resulta válido considerar la diferencia entre eventos agudos y crónicos sobre el deterioro en la capacidad de respuesta biológica frente a estas injurias (IC aguda versus IC crónica). Al respecto, previos reportes estudiaron dichas diferencias y demostraron –en fase crónica– que el aumento de la peroxidación lipídica a través de niveles más altos de TBARS y el aumento del estrés oxidativo produjeron una reducción de la actividad antioxidante total y una mayor actividad de NADPH oxidasa. Esto fue demostrado por Rinaldi Tosi et al., y que además se acompañó de una disminución de la expresión de la isoforma inducible de Hsp70101. Por lo tanto, mayores estudios deberán conducirse con especial atención en IC y modelos relacionados para profundizar la comprensión de estas vías de señalización alteradas.
FinanciaciónEste trabajo fue económicamente apoyado por subsidios para investigación de la Secretaria de Ciencia, Técnica y Postgrado de la Universidad Nacional de Cuyo, Mendoza, Argentina; y por la Agencia Nacional de Promoción Científica y Tecnológica de la República Argentina, Ambos, otorgados al Dr. Walter Manucha (PICT 0234-BID 2777 OC/AR).
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.