




As a prevalent cardiovascular disease, heart failure is one of the leading causes of morbidity and premature mortality. Therefore, there is a special interest in the study of efficient markers associated with risk and/or prediction of cardiovascular events. Multiple candidates are proposed, especially those involved in oxidative and inflammatory processes typical of cardiovascular disease, such as superoxide anion, nitric oxide, and peroxynitrite. There is a lack of knowledge on the potential usefulness of these systems as biomarkers. This review aims to contribute to a better understanding of these systems, as well as an improved patient profile. Furthermore, a deep knowledge of these complex systems would also allow proposing new lines of research for the development of new therapeutic tools as a promising start for new approaches to this disease.
Como enfermedad cardiovascular prevalente, la insuficiencia cardíaca es una de las principales causas de morbimortalidad prematura. Por ello, existe un especial interés sobre el estudio de marcadores eficientes asociados al riesgo y/o predicción de eventos cardiovasculares. En consecuencia se proponen a múltiples candidatos, pero sobresalen especialmente aquellos implicados en procesos oxidativos e inflamatorios propios de la enfermedad cardiovascular como el anión superóxido, óxido nítrico y peroxinitrito. En este sentido, existe una falta de conocimiento sobre las potenciales utilidades de estos sistemas como biomarcadores. La presente revisión procura contribuir a la mayor comprensión de estos sistemas para una mejor caracterización de pacientes. Por otra parte, un profundo conocimiento de estos complejos sistemas también permitiría proponer nuevas líneas de investigación para el desarrollo de inéditas herramientas terapéuticas como una auspiciosa frontera para el abordaje de esta patología.
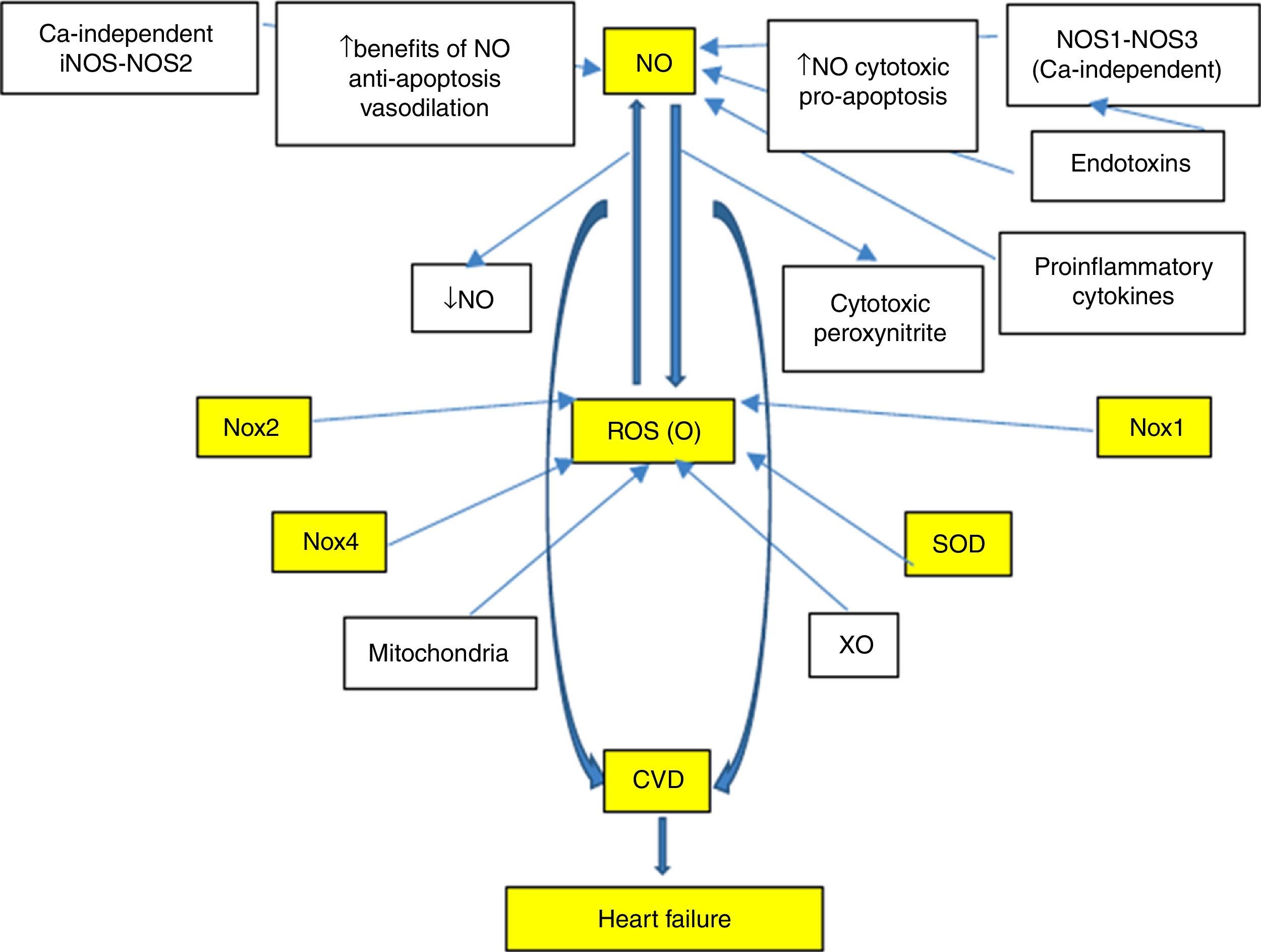
With regard to cardiovascular disease (CVD), there is a special interest in the study and development of efficient markers associated with the risk and/or prediction of events, which in turn make timely intervention possible, finally validating their cardiovascular risk prediction potential. This led to the search for new biomarkers, with the proposal of multiple candidates. Nevertheless, those biomarkers involved in oxidative and inflammatory processes typical of CVD are especially noteworthy. In fact, reactive oxygen species (ROS), especially superoxide anion (O−), and reactive nitrogen species (RNS), such as nitric oxide (NO) and peroxynitrite, show important functions (Fig. 1). In this sense, and with special emphasis on understanding the signalling pathways involved in the pathophysiology of heart failure (HF), there is a lack of knowledge on the potential usefulness of these systems as biomarkers. A better understanding would allow better characterisation of these systems and would also open up new lines of research for the development of novel therapeutic tools, which could mean a promising start for new approaches to this disease.

In particular, various neurohormonal systems have been studied in order to identify biomarkers with good predictive capacity; however, only a few meet all the criteria required to be useful in clinical practice. It is therefore necessary to continue searching for more and better substances that make an improved contribution.
In recent years, CVD has been recognised as a continuum that involves multiple entities, such as primary disease of the heart muscle (cardiomyopathy), hypertension (HTN), left ventricular hypertrophy (LVH), atherosclerotic heart disease, cardiac arrhythmias and diabetes mellitus; and HF is the final common pathway of all these entities with alteration of the signalling pathways involving NO, ROS/RNS, NADPH oxidase (Nox) and superoxide dismutase (SOD).
HF is often tackled from the perspective of the main mechanisms that induce ventricular damage and remodelling as a result of neurohormonal overstimulation by the renin-angiotensin-aldosterone (RAAS) and adrenergic systems. These alterations are classic and mark the progression of the disease. Of particular interest for this review is the fact that increased peripheral vascular resistance and cardiac remodelling constitute the main alterations and are associated with the signalling pathways mentioned above. Therefore, evaluation of HF development from a new perspective for clinical practice is a novel proposal, such as remodelling via the signalling pathways involving NO, Nox and SOD. According to this suggestion, it has recently been reported that damage to the signalling pathways involving NO and related factors may be associated with prognosis and/or mortality during HF.
However, the use of classic biomarkers in HF and drugs for its treatment are expensive and hard to obtain, especially within the field of public health. Consequently, HF is a disease of epidemiological, health and economic importance. This justifies the support offered for major research efforts to ensure a better understanding, treatment and follow-up. The development of new, more accessible and more affordable methodologies for studying its progression/prognosis would therefore allow the natural history of HF to be positively altered. Implementation of these types of biomarker, which are used to improve life expectancy and quality of life, may also help studies on the evaluation of drugs and electrophysiological devices during HF.
Biomarkers in heart failureIn general terms, biomarkers provide useful prognostic information in HF patients and there is currently considerable interest in determining the ability of biomarkers to guide therapy in cases of acute and chronic HF. Under these precepts, Richards and Braunwald listed neurohormonal, inflammatory, oxidative stress, interstitial matrix remodelling, myocyte injury and other newer markers that reflect different pathophysiological aspects of HF.1,2
Neurohormonal markers known as B-type natriuretic peptides (BNP, NT-proBNP and proBNP) are currently preferred due to their diagnostic/predictive value. These are secreted in both the left ventricle (LV) and right ventricle (RV) and specifically reflect LV filling pressure and parietal stress. However, they also increase when the patient has RV dysfunction in the absence of LV dysfunction, as is observed, for example, in respiratory diseases with chronic cor pulmonale and in group 1 patients with pulmonary arterial hypertension (Nice). These markers allow HF to be ruled out with reduced inaccuracies and costs and serial BNP testing is a useful tool for identifying the best discharge time and post-discharge progression.
In view of the above, biomarkers are generally useful, but they also have limitations at the time of use. Their clinical usefulness is related to the fact that testing allows for clinical management and improves prognosis for one or more situations and consequently improves diagnostic certainty associated with risk of onset or worsening of HF (the ideal thing is to have a response with a specific treatment). Monitoring through serial testing of markers should also improve the results obtained during patient follow-up, i.e. less acute decompensation, reduced mortality and/or improved quality of life.
However, the use of biomarkers in HF questions the role of the laboratory and the value of a simple blood sample for diagnosis, prognosis, monitoring of progression and to guide therapy. Moreover, an improved knowledge of the importance of neurohormonal systems in HF progression has resulted in major therapeutic advances since the mid 1980s. This has been based on the study of circulating concentrations of myocyte stress markers, BNP, proBNP and NT-pro BNP, which were also closest to the typical ideal biomarker. Nevertheless, in addition to the aforementioned markers, special attention has been paid over recent years to investigating oxidative stress modulators and NO, Nox-ROS and SOD-mediated signalling pathways that exhibit great involvement in the development of CVD (Fig. 1). A profound knowledge of such matters will help us gain a better understanding of the mechanisms, neurohormonal changes and biomolecular damages that have not yet been well established and that occur during HF.
Nitric oxide as a biomarker in heart failureMultiple studies have shown the involvement of NO in the pathophysiology of HF and that there is a marked NO/redox disequilibrium at the expense of increased free radical production by enzymatic pathways, including especially vascular Nox, cardiac XO, mitochondrial enzymes and haemoglobin oxidase in red blood cells. In general terms, this oxidises proteins critical for excitation-contraction coupling and also diminishes NO bioavailability at the expense of altering the activity and/or site of producing enzymes (NOS and XO).3,4 This leads to the characteristic mechanoenergetic uncoupling since reduced contractility is not accompanied by a proportional reduction in energy consumption. There is also marked neurohormonal activation with increased proinflammatory cytokines that induce NOS2 expression5 and sometimes increase NOS1 and NOS3 activity, modifying the site of NOS1.6,7 However, the hypothesis that NO plays a major role in the aetiopathogenesis of HF does not match the finding that the same element exerts a cardioprotective effect during ischaemia or that there is reduced NOS expression in failing hearts and/or impaired bioavailability in patients with HF. In fact, in experimental models and in patients with HF, NOS3 activity may increase, decrease or remain the same,4,8,9 and NOS2 has been observed to increase dramatically in some studies, but not in all cases.5–7,10–14 This would suggest, at least in part, that changes in NO2 and NOS3 expression may be due to an epiphenomenon that accompanies HF, without being determinants of its cause (Fig. 1).
In failing hearts, decreased NO would lead to reduced endothelium-dependent coronary vasodilation, impaired ventricular relaxation and increased maximal oxygen consumption (MVO2).15,16 Therefore, NOS3 overexpression should improve these alterations as shown in some experimental studies. Also, after coronary artery ligation, there is enhanced contractile performance and reduced LV hypertrophy and mortality.17–19 However, NOS3 knock-out mice that also received coronary artery ligation showed LV hypertrophy and dilation, decreased fractional shortening, lower ejection fraction, greater end-diastolic volume, LV internal diameter and vascular rarefaction and higher mortality.8,20 Enalapril and valsartan also had less beneficial effects in these mice.21 These results suggest that NOS3 reduces ventricular dysfunction and post-infarction remodelling and may also play some sort of role in angiotensin II-induced ventricular dysfunction.
However, it has been reported in patients with dilated cardiomyopathy (DCM) that decreased NO does not alter cardiac contraction, and with intracoronary infusion of sodium nitroprusside or substance P, it does not modify haemodynamic parameters.10,22–24 Nevertheless, one animal model with pacing-induced HF was able to show that the cardiac decompensation phase was characterised by a marked reduction in NO production, dP/dtmax and ventricular distensibility (which increases LV end-diastolic pressure with elevated MVO2), while cardiac metabolism switched from using fatty acids to using glucose as an energy source15; these results suggest that NO may participate in the coupling of coronary flow, contractile performance and cardiac metabolism.
Patients with DCM have increased NOS1 expression, which is translocated from the sarcoplasmic reticulum (where it is coupled to XO) to the sarcolemma, where, as in the case of NOS3, it interacts with a calmodulin-regulated calcium-dependent ATPase.7 These alterations in expression and site may be beneficial during HF since NOS1: (a) inhibits the heart's response to β-receptor stimulation via inhibition of the Ca2+ current, and Ca2+ release from the sarcoplasmic reticulum may perhaps induce cardioprotective effects against catecholamine-induced cardiotoxicity25; (b) increases cardiac vagal tone, decreasing heart rate26; (c) restores central baroreceptor activity; and (d) may compensate for NOS3 inhibition.7 However, it is also possible that translocation of NOS1 to the sarcolemma may facilitate oxidative stress since its control on XO activity in the sarcoplasmic reticulum would be lost and this would alter the nitric oxide/redox balance at the sarcolemma.9
It is interesting that, during HF, reduced inotropic response to β-stimulation is observed as a result of alterations in receptor density (with decreased β1 and β2 receptors and increased β3 receptors which mediate negative inotropic responses) or in coupling of the receptor to its signalling pathways (with increased expression of β-adrenergic receptor-specific kinase or ARK and Gi proteins and decreased Gs proteins). The β3 receptors are more resistant to homologous desensitisation due to the increased sympathetic tone that is characteristic of HF and their stimulation may therefore facilitate continuous NO production in the presence of increased sympathetic tone in the setting of HF.27 Therefore, NO synthesised through the β3-NOS3 pathways, NOS1 translocated to the sarcolemma and the induction of NOS2 may modulate response to catecholamines and antagonise their toxicity in the failing myocardium. In fact, inhibition of NOS potentiates increased contractility produced by β-adrenergic agonists in animal models16 and in patients with HF.9,28,29 However, it is logical to assume that NO is only one of the factors to regulate response to β-adrenergic agonists in HF patients.30 Nevertheless, under certain circumstances, an increase in synthesised NO following induction of NOS2 may be beneficial since it improves ventricular relaxation,6,22 reduces MVO231 and response to β-adrenergic stimulation8,11,15 and increases angiogenesis.32 In cardiomyocytes of heart transplant patients, isoproterenol produces a slight increase in contractility and heart rate, and, where NOS2 inhibition normalises both responses, it also increases Ca2+ transport. However, NOS2 inhibition has no effect on normal cardiomyocytes or on cardiomyocytes of HF patients in whom response to isoproterenol was preserved and in whom NOS2 expression is poor.14 In other words, in HF patients, NOS2 expression limits response to β-adrenergic agonists, an effect that may be mediated by inhibition of Ca2+ release from the sarcoplasmic reticulum via a cGMP-independent pathway and which is related to alterations in cellular redox status produced by peroxynitrite.33 Cardiac-specific NOS2 overexpression in mice leads to increased production of peroxynitrite and dilatation, hypertrophy and cardiac fibrosis; moreover, although they rarely develop HF, they display a high incidence of sudden cardiac death due to bradyarrhythmia.34 On the contrary, mice with NOS2 overexpression display a normal genotype since the NO produced is neutralised by cytoplasmic myoglobin; however, when these experiments are repeated in myoglobin-deficient mice, the animals develop signs of hypertrophy, interstitial fibrosis and ventricular dilatation.35
As mentioned above, during HF, enzyme pathways that produce free radicals are upregulated and NO-producing enzymes (NOSs and XO) are altered, producing vasoconstriction and mechanoenergetic uncoupling. In fact, in animal models, the transition to decompensated HF implies NOS1 and NOS3 deficiency and consequently poor cardiac NO synthesis, plus increased XO activity.4 NO also regulates a Nox that inhibits Ca2+ release from the sarcoplasmic reticulum and, therefore, the balance between NO and oxidative stress also regulates cardiac function through its effects on intracellular Ca2+ cation signalling. XO, an important source of superoxide radical, increases in the myocardium and vessels of patients with HF,36 producing endothelial dysfunction, depressed cardiac function, mechanoenergetic uncoupling and apoptosis.37,38 XO inhibition with allopurinol improves mechanical efficiency, post-infarction myocardial remodelling and response to catecholamines.39 It is important to note that the effects of allopurinol are suppressed by blocking NOS with L-NAME (specific inhibitor) and the effects of this inhibitor are blocked with allopurinol,38 which indicates that there may be interaction between the two signalling pathways.4 However, some drugs used in HF, such as statins40 and ACE inhibitors,41 alter the NO/redox balance by increasing bradykinin levels and NO synthesis and reducing the production of free radicals (superoxide, peroxynitrite) upon inhibiting NADPH oxidase (Fig. 1).
NO also plays a major role in cardiac ion channels, the genesis of cardiac arrhythmias, cardiac apoptosis, ischaemic pre-conditioning, cardiac ischaemia and mitochondrial function. The damage or benefit modulated by NO will depend on cell type and condition. Therefore, high concentrations induce cell death during ischaemic injury leading to neurodegenerative diseases.42–44
Cytotoxicity attributed to NO is due to peroxynitrite produced by the diffusion-controlled reaction between NO and the superoxide anion. Peroxynitrite interacts with lipids, DNA and proteins via direct oxidative reactions or indirect radical-mediated mechanisms. These reactions trigger cellular responses ranging from subtle modulations of cell signalling to overwhelming oxidative injury leading to cell necrosis or apoptosis. Peroxynitrite is crucial in conditions such as stroke, myocardial infarction, chronic heart failure (CHF), diabetes, cardiogenic shock, chronic inflammatory diseases, cancer and neurodegenerative disorders.45
The cardioprotective effect responds at least partly to the protein kinase G pathway (GMP/PKG),46,47 where NO activates a soluble guanylate cyclase that catalyses cGMP synthesis from GTP. Furthermore, NO participates in mechanisms with PKG-mediated anti-apoptotic actions and these are an active area of research as modulation of these pathways may have major therapeutic implications.
Nitric oxide-related factors as biomarkers in heart failureIn patients with chronic heart failure, the increased production of ROS, and in particular the importance of O−, have been clearly demonstrated. Evidence shows the role of both ROS and RNS in the pathophysiology of CVD. Consequently, multiple biomarkers of oxidative stress common to both systems are evaluated. Of these, Nox is the most efficient system and represents a major source of ROS in HF. Abnormal activation of the renin-angiotensin system (RAS), which significantly affects the induction of apoptosis and fibrosis, is a result of the production of ROS by Nox and is specific to CVD.48,49 In this respect, the emphasis is on changes in the ROS-Nox-dependent signalling pathways as significant factors responsible for the development of many cardiac diseases. NADPH oxidases (Nox) are major sources of O− in vascular cells and myocytes, where they share some of the characteristics of the neutrophil enzymes. In response to growth factors and cytokines they produce O−, which may be metabolised to hydrogen peroxide. Both of these reactive oxygen species act as second messengers to activate multiple intracellular signalling pathways. In addition to Nox from myocytes, a vascular Nox has been found to be essential in the physiological response of vascular cells, including growth, migration and modification of the extracellular matrix. They have also been linked to HTN and disease states associated with uncontrolled growth and inflammation, such as atherosclerosis. In the case of pressure overload, myocardial Angiotensin II (Ang II) production is activated, which stimulates intracellular signalling pathways which in turn activate hypertrophic response, as is the case with Nox.50
Nox has various constituents or subunits which are relevant for its relationship with CVD, shown in order of importance as Nox2, Nox4 and Nox1 and the subunits gp91phox, p22phox, p47phox and Rac1. These play an important role in cardiac damage, causing cardiac hypertrophy and opposite cellular responses, accelerating atherosclerosis, hypertension and myocardial remodelling and activated in HF and post-AMI.50–52
More specifically, Esposito et al.53 evaluated mice from which Rac1 had been eliminated, and came to the conclusion that this prevented Ang II-induced hypertrophy. Rac1 initiated the ROS-dependent hypertrophic response generated by Nox in the heart,54 confirming that the production of O− by the Nox2 subunit Rac1 initiated activation of protein kinase B (Akt) as a component of this signalling pathway and Ang II-induced cardiomyocyte hypertrophy.55
Basic research provides evidence for Nox2 being responsible for the vascular production of ROS, lower NO bioavailability and the development of early lesions in mice aortas.56 An additional finding combines the involvement of circulating transforming growth factor-beta (TGF-β) and apolipoprotein E, where the increase of TGF-β induced Nox activation and overproduction of ROS, accelerating atherosclerosis, hypertension and myocardial remodelling in apolipoprotein E-deficient mice.57
However, elevated expression of Nox and O− were also described in the carotid arteries of rabbits with CHF58 and specifically in the activity of the Nox2 subunit p47phox in the LV of mice after myocardial infarction (MI).59
While Nox2 is involved in Ang II-induced LVH, Nox4 is apparently also involved in pressure overload in mouse myocardium.60 Accordingly, this isoform may also play an important, although controversial, role in CVD. It has been suggested that Nox4 was bound to the p22phox protein on the internal membrane of epithelial cells. It was also found—in contrast to other NADPH oxidase isoforms—that Nox4 mainly produced hydrogen peroxide and a very small quantity of O−. Cytosolic oxidase proteins or the GTPase Rac were not required for the activity of this enzyme.61 Accordingly, there is some evidence mentioning that although Nox4 produced hydrogen peroxide, it also generated O− intracellularly.62 However, there is still considerable uncertainty regarding the production of Nox4-dependent ROS. Therefore, the current state of knowledge in this regard indicates that Nox4 primarily produces hydrogen peroxide and not O−, contradicting most other experimental data. A possible explanation would be the use of unreliable methods such as nitro blue tetrazolium reduction to detect O−.62,63 However, the use of specific and precise methods such as chemiluminescence to detect O− provided different results.64 For example, with the lucigenin chemiluminescence assay it was found that Nox4 and Nox2 produced approximately 75% of O− in the coronary arteries of patients with coronary artery disease.65
Nox4 is located at various sites within cardiac cells compared to other Nox. In particular, it is located in the mitochondria where it represents a major source of O− production in myocytes. It can bring about mitochondrial dysfunction, cell death, left ventricular dysfunction in response to pressure overload and, paradoxically, it may also cause cardiac adaptation to chronic stress.66 In aged mice under hypertrophic stimulation with pressure overload, Nox4 was stimulated and this increased the production of O− and induced cardiac dysfunction with fibrosis and apoptosis.67
Unlike other NADPH oxidase isoforms, Nox4 stimulation in cardiomyocytes caused protection against pressure overload-induced cardiac remodelling. The authors justify these results because of the preservation of Nox4-induced myocardial capillary density through activation of hypoxia-inducible factor-1 (Hif1) and the production of vascular endothelial growth factor. They also showed that the site of Nox4 in cardiomyocytes was not mitochondrial, but in the perinuclear endoplasmic reticulum. These findings contradict those of other authors.66 There are contradictory results concerning the effects on oxidative stress and its localisation. Further research is required regarding the origin of these differences68 because they conflict with most findings on the effects of Nox in cardiomyocytes and other cells.
Unlike the implications of Nox in cardiac diseases, the involvement of XO in ROS-dependent cardiac damage originally raised many questions. XO and xanthine dehydrogenase (XDH) are the oxidised and reduced forms of xanthine oxidoreductase (XOR). XO was considered the greatest source of O− and hydrogen peroxide, and its mechanism was initially well established.63 Paradoxically, subsequent studies discovered low XO activity in animal and human hearts.69,70
Currently, however, there is consensus on the increase in XO levels and their activity in the cardiovascular system under pathological conditions, but that they cannot be detected easily in physiological conditions. Consequently, Thompson-Gorman and Zweier measured the production of XO-mediated ROS in isolated rat heart.71 They found that XO was an important factor of oxidative damage from ischaemia/reperfusion in the rat heart. Accordingly, it has also been demonstrated that the increased production of XO-catalysed ROS during ischaemia/reperfusion results from increased concentration of the substrate (xanthine and hypoxanthine) on account of ATP degradation during ischaemia.72 Ashraf and Samra also suggested that XO activity increases during ischaemia and was intensified following reperfusion.73 XO is found in interstitial cells, coronary vascular endothelium and smooth muscle cells. De Jong et al.74 showed that the production of ROS by xanthine oxidoreductase (XOR) increased in HF, but not in cardiac hypertrophy.
Like the NADPH oxidases, XO led to many ROS-dependent cardiac disorders such as endothelium-mediated deterioration of vasodilatation, increased XO activity in dilated cardiomyopathy, NO reduction in patients with coronary disease, coronary endothelial dysfunction and toxic effect of O− caused by XO in the heart. Moreover, the increased activity of XO and the reduction of extracellular superoxide dismutase (ecSOD) caused deterioration of endothelium-mediated vasodilatation in patients with HF.75 To that effect, later research established levels of protein XO and XO-dependent O− induced by Ang II in endothelial cells of patients with coronary disease,76 thereby suggesting that Ang II promotes overproduction of O− by activation of redox-sensitive XO. In addition, basic studies confirmed the role of XO, as it was discovered that its activity would be elevated in DCM77 and that chronic XO inhibition by allopurinol suppressed the progression of HF to DCM.
Rats with HF on account of spontaneous hypertension also showed increased mRNA expression and XOR activity, while XOR inhibition caused a reversal of remodelling in HF on account of spontaneous hypertension and dilated cardiomyopathy.78 XOR and Nox also increased cardiac O− formation in Dahl salt-sensitive hypertensive rats with diastolic HF.79
XO-produced ROS reduced the bioavailability of coronary NO in patients with coronary artery disease (CAD).80 Similarly, in a mouse model, myocardial ischaemia/reperfusion increased the expression of tumour necrosis factor-alpha (TNF-α) and induced XO activation and O− production bringing about coronary endothelial dysfunction.81 Moreover, another toxic effect of XO-generated O− was demonstrated in the hearts of spontaneously hypertensive rats with HF with DCM. Specifically, O− caused deterioration of S-nitrosylation of the ryanodine receptor (RyR) resulting in calcium leak from the sarcoplasmic reticulum in skeletal muscle.82
Of particular interest and widely recognised, mitochondrial dysfunction is an essential source of ROS in CVD, and consequently mitochondria are the subject of ongoing study. It has been established that O− generated by the mitochondria results in the release of transporter electrons from the respiratory chain (complexes I and III).83 Many authors have confirmed the importance of the overproduction of mitochondrial ROS in the failing heart (myocardial insufficiency, ischaemia, ischaemia/reperfusion, coronary endothelial dysfunction caused by O− in congestive HF, progression of LVH to pulmonary hypertension (PH) and right-sided HF, mitochondrial and LV damage and, finally, arrhythmias).
Originally, mitochondrial complex I was described as a potential source of ROS in the myocardium of dogs with HF.84 Moreover, ischaemia increased ROS production in isolated mouse heart mitochondria.85 The production of mitochondrial O− in the failing heart can also induce complex II changes in the post-ischaemic myocardium of rats subjected to coronary ligature followed by reperfusion.86 It should be noted that damage caused by ischaemia/reperfusion with the ablation of protein p66 (Shc) in mouse hearts plays a key role in the formation of mitochondrial ROS.87 In that regard, mitochondrial depolarisation and the increased production of lipoxygenase-mediated ROS and arachidonic acid induced arrhythmias due to ischaemia/reperfusion.88 In addition, the increased production of mitochondrial O− was responsible for coronary endothelial dysfunction and reduced coronary flow in congestive HF.89
It should be emphasised that studies performed during the progression of LVH to congestive HF demonstrated that mitochondrial Nox was the primary source of ROS—especially in hypertension-induced right ventricular failure. Surprisingly, the increased activity of mitochondrial complex II was the mechanism responsible and not of complexes I and III, acknowledged as the greatest sources of mitochondrial ROS. This contribution was particularly relevant for the production of ventricular ROS in HF.90 However, Mariappan et al.91 demonstrated that the production of mitochondrial O− induced by TNF-α increased complex I activity of the respiratory chain which caused mitochondrial damage in rat LV. All these findings suggest that the overproduction of ROS by the mitochondria represents a causal origin in cardiac diseases.
Based on the above, it is clear that CVD is associated with a chronic state of oxidative stress and inflammation mediated by complex, interconnected signalling pathways. More specifically, the course of HF is characterised by mitochondrial dysfunction, overproduction of ROS, activation of RAS linked to greater activity of NADPH oxidase and NO reduction (Fig. 1). Interestingly, it has been demonstrated that lower NO bioavailability induces heat shock protein (Hsp70) expression, which causes beneficial effects against oxidative stress damage, inflammation and apoptosis. The induction of heat shock proteins as a response to damage by stimulation of the RAS system and/or NO deficiency was originally suggested by Bravo et al.92
Currently, it is understood that heat shock proteins are elevated in the plasma of patients with CVD. However, their physiological role is not yet fully understood and their value for predicting the development and/or progression of the disease is understood even less. In particular, it was suggested that circulating levels of Hsp70 could indicate the presence/progression of atherosclerosis in subjects with established hypertension; in addition, an intriguing possibility posed by the authors is that Hsp70 could protect against oxidative and inflammatory damage in this group of subjects.93 Accordingly, it has recently been proposed that Hsp70 could be a potential biomarker and treatment objective in diseases such as cancer, CVD and neurological and hepatic diseases.94
However, there is information indicating that low Hsp70 levels may be related to a healthy cardiovascular status, and it was suggested as a predictor of longevity.95 Based on this, Hsp70 was investigated in patients with CHF in an attempt to re-establish a relationship between severity and survival. Elevated Hsp70 levels, particularly in those subjects with cardiac cachexia, were found and, therefore, could be related to disease severity, although not to survival.96 Consequently, further study is required to understand the significance of the relationship between Hsp70 expression and CHF morbidity.
Conclusions and perspectivesHF is, in almost all cases, the end result of primary heart disease and any cause of structural damage to the heart, and is one of the most prevalent CVD. This makes it one of the main causes of premature morbidity and mortality in most developed and developing countries. Of particular interest is the fact that evidence obtained from sources such as prospective and interventional epidemiological cohort studies suggest that this disease is associated with alterations in endothelial function, oxidative metabolism, inflammation and apoptosis. Some of the most relevant determinants of such alterations include SOD activity, phosphorylation and expression of NO-producing enzymes, increased glutathione peroxidase activity, activation of NADPH oxidase and expression of p22phox.
As a result, the literature highlights that HF patients display alterations in signalling pathways that promote oxidative stress and inflammation. Such alterations include specifically elevated ROS with increased NADPH activity and reduced SOD activity. This was recently proven by our laboratory.97 However, in this same context, low NO levels were also reported. These may be due to interactions with ROS, decreased NO production or a combination of the two. Although there are many basic research references on alterations of NO, Nox-ROS and SOD pathways, there are few references regarding their actual clinical impact. In spite of current knowledge and it now being more than 20 years since the functions of endogenous NO were identified, attempts to generate new therapeutic strategies have not been fruitful, reflecting slow progress in the study and understanding of potential related biomarkers.
Alteration and/or uncoupling of Nox, ROS and SOD conditions multiple signalling pathways involved in LVH, myocardial failure, progression of HF to right-sided heart failure, altered endothelial function and coronary artery vasomotility and reperfusion injury and arrhythmia, all components of the cardiovascular continuum. In this context, NO is a key component for maintaining cardiovascular homeostasis, and impaired NO bioavailability is therefore clearly associated with CVD. There is substantial evidence, as outlined in this review, that ROS generated from NADPH oxidase may be primarily responsible for reduced NO bioavailability. Thus, O− may interact with NO to produce peroxynitrite, which may in turn give rise to other reactive species. Specifically, increases in oxidative stress during HF may be the result of the functional uncoupling of the respiratory chain due to impaired antioxidant capacity (reduced SOD activity and/or stimulation of enzymatic sources, including NADPH oxidases).
The proposal to evaluate NO as a biomarker in HF, as proposed by our working group and others, responds to the assumption that this factor is involved in relevant pathophysiological aspects of HF. In fact, there is NO/redox disequilibrium in HF with increased enzymatic pathways that produce free radicals (NADPH oxidases, cardiac XO, mitochondrial enzymes, haemoglobin oxidase in red blood cells, etc.) which oxidise proteins involved in the excitation-contraction coupling and inactivate NO and alter the activity and site of NO-producing enzymes. This leads to functional uncoupling characterised by reduced contractility that is not accompanied by a similar reduction in energy consumption. This also conditions marked neurohormonal activation and increased proinflammatory cytokines. In this sense, it has been suggested very recently that low NO bioavailability induces the expression of oxidative stress proteins, such as Hsp70, which promotes protective effects against damage not only from oxidative stress, but also from inflammation and apoptosis. In fact, our laboratory suggested that Hsp70 may be used as a potential biomarker of oxidative-inflammatory damage.98 Consequently, recent studies suggest that heat shock proteins play a key role in the pathogenesis of cardiovascular diseases, including HF. In this regard, it is unclear whether circulating Hsp70 levels are related to CVD risk factors, echocardiographic indexes of LV remodelling and/or prevalence of CVD.99 However, the analysis of preliminary results from our laboratory suggests that patients with HF also have elevated inflammatory markers (C-reactive protein, IL-6 and TNF-α), while Hsp70 was reduced compared to healthy patients. As a result, the determination of oxidative stress markers, such as plasma inflammatory markers, allowed us to distinguish healthy patients from patients with HF. Such findings of lower plasma Hsp70 levels in patients with HF and with low NO levels were previously unpublished and controversial. Interpretation of our results has proven even more complex since learning about the opposite effects that Hsp70 has at extracellular versus intracellular locations upon activation of the inflammatory pathway by NF-κB.100 However, in relation to oxidative stress and inflammation, it is valid to consider differences in consequences of acute and chronic events on biological response capacity to these injuries (acute HF versus chronic HF). In this regard, earlier reports studied these differences and showed (in the chronic phase) that enhanced lipid peroxidation through higher TBARS levels and increased oxidative stress resulted in reduced total antioxidant activity and enhanced NADPH oxidase activity. This was demonstrated by Rinaldi Tosi et al. and was also accompanied by decreased inducible Hsp70 isoform expression.101 Therefore, more studies should be conducted with special attention paid to HF and related models in order to gain a deeper understanding of these altered signalling pathways.
FundingThis article was funded by research grants from the Department of Science, Technology and Postgraduate Studies of Universidad Nacional de Cuyo, Mendoza, Argentina and from the Agencia Nacional de Promoción Científica y Tecnológica (National Agency for Scientific and Technological Promotion) of the Republic of Argentina, both granted by Dr Walter Manucha (PICT 0234-BID 2777 OC/AR).
Conflicts of interestThe authors declare that they have no conflicts of interest.
Please cite this article as: Bonafede R, Manucha W. Óxido nítrico y factores relacionados a oxidación e inflamación como posibles biomarcadores de insuficiencia cardíaca. Clin Investig Arterioscler. 2018;30:84–94.