




La angiotensina II, el péptido efector del sistema renina-angiotensina, está implicado en la patogénesis de la aterosclerosis a distintos niveles. Existen numerosas evidencias experimentales que demuestran que tanto la inhibición de la síntesis de angiotensina II mediante la administración de inhibidores de la enzima de conversión de la angiotensina II como mediante el empleo de antagonistas de su receptor AT1 inhiben la formación y la progresión de la lesión aterosclerótica. La angiotensina II es capaz de estimular la producción de especies reactivas de oxígeno en el vaso que desempeñan un papel clave en la disfunción endotelial y en la oxidación de las lipoproteínas de baja densidad (LDL). Asimismo, la angiotensina II participa en la inducción de la respuesta inflamatoria en la pared vascular mediante la producción de moléculas de adhesión y citoquinas quimiotácticas y proinflamatorias. Este péptido estimula la proliferación y la migración de células de músculo liso y modula su cambio fenotípico, dando lugar a un aumento en la síntesis de la matriz extracelular. Finalmente, la angiotensina II también participa en las complicaciones de la aterosclerosis al favorecer la ruptura de la placa y la trombogenicidad de la misma. Por tanto, la angiotensina II juega un papel importante tanto en el inicio del proceso al favorecer la disfunción endotelial, en la progresión de la lesión ateromatosa, en la ruptura de la placa y en la aparición de accidentes trombóticos.
Angiotensin II, the effector peptide of the renin-angiotensin system, may be involved in various factors affecting the pathogenesis of atherosclerosis. There is abundant experimental evidence that both pharmacological antagonism of angiotensin II formation by angiotensin converting enzyme inhibition and blockade of angiotensin II by angiotensin type I receptor blockade inhibits the formation and progression of atherosclerotic lesions.
Angiotensin II is able to stimulate the production of reactive oxygen species in blood vessels, which play a key role in endothelial dysfunction and oxidation of low-density lipoproteins. In addition, angiotensin II participates in the induction of the inflammatory response in the vascular wall through the production of adhesion molecules and chemotaxic and proinflammatory cytokines. This peptide stimulates the proliferation and migration of smooth muscle cells and modulates phenotypic changes in these cells, thus increasing the synthesis of extracellular matrix. Finally, angiotensin II also contributes to the complications of atherosclerosis by favoring plaque rupture and thrombogenicity. Therefore, angiotensin II plays an important role both in the beginning of the process —promoting endothelial dysfunction— in atherosclerotic lesion progression, in plaque rupture, and in the occurrence of thrombotic accidents.
Durante el proceso de aterogénesis se dan una serie de cambios a nivel vascular en los que las células inflamatorias, en particular los monocitos, los macrófagos y los linfocitos, son piezas clave. Asimismo cabe destacar el papel del endotelio en dicho proceso inflamatorio, así como la importancia de la acumulación de partículas de colesterol LDL oxidadas en la íntima que contribuyen al reclutamiento de monocitos y su transformación a células espumosas1.
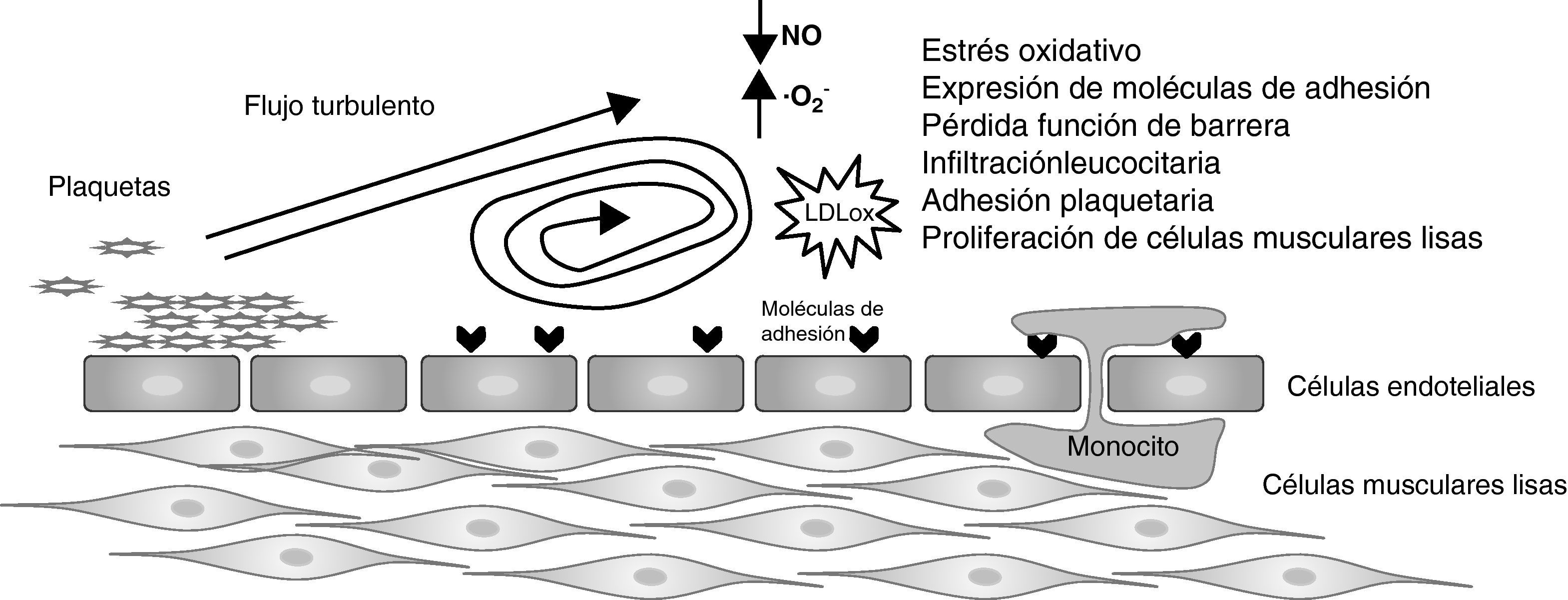
En el inicio de la lesión, el endotelio cobra especial relevancia por su capacidad de generar moléculas efectoras que regulan el tono vasomotor, la agregación plaquetaria y la coagulación, y previenen la adhesión de las células sanguíneas, la inflamación y el remodelado vascular. Uno de los primeros acontecimientos del proceso aterosclerótico es la acumulación de LDL en la matriz subendotelial. Estas partículas difunden libremente entre las uniones celulares y su retención en la pared vascular se debe principalmente a la interacción entre la apolipoproteína b (ApoB) constituyente de las LDL y los proteoglicanos de la matriz extracelular2. Las partículas de LDL deberán experimentar una serie de modificaciones, entre las que se incluyen oxidación, lipólisis, proteólisis y agregación, antes de ser reconocidas por los receptores scavenger de los macrófagos. La acumulación de partículas LDL oxidadas estimula la producción por parte de las células endoteliales de moléculas proinflamatorias (IL-1, IL-6), moléculas de adhesión (ICAM, VCAM) y factores de crecimiento como el factor estimulante de macrófagos (M-CSF), que estimula la proliferación y la diferenciación de macrófagos así como la expresión de receptores scavenger. Todos ellos favorecen la adhesión, la migración y la acumulación de monocitos y linfocitos T en el espacio subendotelial3. La transformación de los macrófagos tisulares en células espumosas se produce por la captación de LDL oxidadas a través de los receptores scavenger. Esta captación por los macrófagos no está limitada como la de las LDL nativas, que está controlada negativamente por los niveles de colesterol. A medida que progresa la lesión se produce la proliferación y la migración de células musculares debido a la liberación de factores de crecimiento por las células inflamatorias acumuladas en la pared. Además, se produce la transformación de las células del músculo liso de fenotipo contráctil a fenotipo sintético, y la consiguiente formación de matriz extracelular4. Esto determina, finalmente, la remodelación de la pared vascular y la formación de una cápsula fibrosa que recubre el núcleo lipídico. Las regiones vasculares más propensas a desarrollar lesión son las que presentan un flujo discontinuo o turbulento que promueve el desarrollo y la progresión de la aterosclerosis5 (fig. 1). El flujo laminar no sólo es un estímulo para la síntesis y la liberación de óxido nítrico (NO), sino que además reduce la formación de anión superóxido (O2-). Esto genera un efecto antitrombótico, antioxidativo y antiproliferativo en la monocapa de células endoteliales, preservando además su función como barrera selectiva. Por el contrario, las regiones vasculares con lesión o zonas de ramificación presentan un flujo discontinuo y/o turbulento que favorece la producción de anión superóxido en detrimento del NO5.
 y acelera la formación de anión superóxido (O2-). Este puede promover un estado protrombótico, oxidativo y proliferativo en la superficie vascular, promoviendo la infiltración de leucocitos hacia el espacio subendotelial5.")
Inicio del proceso aterosclerótico generado por un flujo turbulento. El flujo turbulento reduce la generación a nivel local de óxido nítrico (NO) y acelera la formación de anión superóxido (O2-). Este puede promover un estado protrombótico, oxidativo y proliferativo en la superficie vascular, promoviendo la infiltración de leucocitos hacia el espacio subendotelial5.
La inflamación desempeña un papel importante no sólo en el desarrollo de la lesión ateromatosa, sino también en la inestabilidad de la placa y la aparición de episodios vasculares agudos como consecuencia de la ruptura de la misma; los macrófagos y linfocitos T son responsable de la liberación de enzimas proteolíticas y moduladores de matriz que debilitan la cápsula y favorecen su posible rotura6. Esta suele producirse en los extremos de la misma que están sometidos a mayor estrés hemodinámico y donde la cápsula fibrosa es más delgada, tiene menor contenido de colágeno y células musculares y mayor contenido de macrófagos y de colesterol7.
Angiotensina II y desarrollo ateroscleróticoEl sistema renina-angiotensina tiene un papel crítico en la iniciación y la progresión del proceso aterosclerótico, y contribuye por lo tanto al desarrollo de la enfermedad cardiovascular. La angiotensina II (AII) se ha reconocido desde hace muchos años como un agente vasoconstrictor tanto local como sistémico, con acciones sobre el volumen extracelular modificando la reabsorción de sodio en los segmentos tubulares distales de la nefrona (directamente o a través de la aldosterona). Mientras en el pasado se creía que la AII afectaba al proceso aterosclerótico a través exclusivamente de su efecto hemodinámico, en las dos últimas décadas se ha demostrado que la AII per se es capaz de acelerar el proceso aterosclerótico8 afectando a los cambios estructurales y funcionales de la pared vascular.
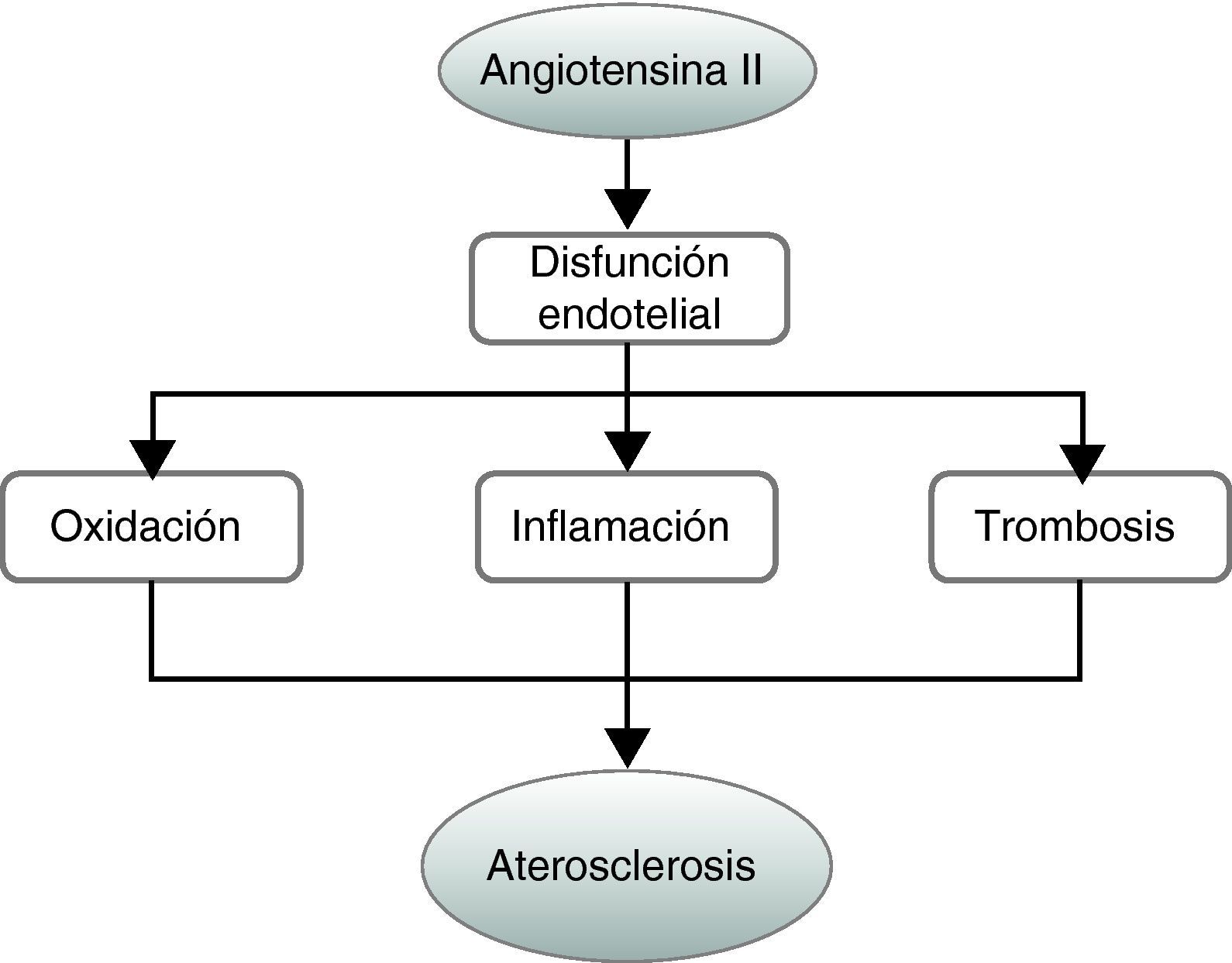
En los estadios iniciales de la aterosclerosis se detecta la existencia de disfunción endotelial, caracterizada por una disminuida vasodilatación dependiente de endotelio y la captación y oxidación de partículas de colesterol LDL en la íntima que desencadena un proceso inflamatorio en la pared vascular. Estos episodios se dan, al menos en parte, por un aumento local de especies reactivas de oxígeno (EROS). La AII desempeña también un papel clave en la respuesta inflamatoria que subyace al proceso aterosclerótico alterando la permeabilidad del endotelio y favoreciendo la infiltración de leucocitos y macrófagos al espacio subendotelial. Este péptido favorece también la progresión de la lesión aterosclerótica porque participa en la migración y la proliferación de las células musculares lisas, así como en su diferenciación hacia un fenotipo sintético, lo que determina la producción de matriz extracelular (colágeno, elastina y proteoglicanos). El sistema renina-angiotensina también puede participar en la inestabilidad de la placa y en la aparición de episodios agudos al favorecer la trombogenicidad de la misma alterando el balance de coagulación y fibrinólisis (fig. 2).

La angiotensina II desempeña un papel clave en el desarrollo de la aterosclerosis al participar en la disfunción endotelial asociada a ella. La alteración de la función endotelial favorece el aumento del estrés oxidativo, la estimulación de la respuesta inflamatoria y la trombosis que subyacen a la aterosclerosis.
A continuación se revisa con más detalle el papel de la AII en cada uno de los aspectos implicados en el inicio y la progresión del proceso aterosclerótico.
Angiotensina II, NADPH oxidasa y estrés oxidativoEl grado de oxidación en el endotelio está controlado principalmente por la interacción entre el NO y O2-. Alteraciones en la producción de NO e incrementos en su degradación por interacción con el O2- se asocian con aterosclerosis, la hipercolesterolemia, la hipertensión, la diabetes y el tabaquismo9. Además, el NO media efectos antiateroscleróticos inhibiendo la proliferación y la migración de las células de músculo liso vascular, la adhesión de leucocitos al endotelio y la agregación plaquetaria10–12. Sin embargo, el NO puede llegar a ser proaterogénico en el caso de que la inflamación crónica característica del proceso aterosclerótico produzca una sobreexpresión de la óxido-nítrico-sintasa (NOS) inducible. Si el NO y el O2- son producidos alcanzando grandes concentraciones, reaccionarán generando peroxinitrito, un potente agente oxidante13. Este radical participa en determinados procesos implicados en el desarrollo del proceso aterosclerótico, tales como la disfunción endotelial, la oxidación de LDL, la inflamación y la apoptosis14.
En situaciones patológicas en las que los niveles de tetrahidrobiopterina son deficientes en relación a la cantidad de NO sintasa endotelial (NOSe), esta enzima puede desacoplarse generando aniones superóxido en lugar de NO, y este fenómeno parece estar directamente implicado en el desarrollo del proceso aterosclerótico. No obstante, la NADPH oxidasa se considera la principal fuente de O2- en el sistema cardiovascular y es una enzima clave en el desarrollo y progresión del proceso aterosclerótico, según se ha descrito en diversos estudios clínicos y experimentales15. Es importante destacar que, en la mayoría de los casos, el incremento en la actividad de la NADPH oxidasa es consecuencia de su aumento en diversos tipos celulares presentes en la pared vascular, tales como células endoteliales, células de músculo liso vascular, fibroblastos e incluso células inflamatorias infiltradas. Una cuestión que queda aún por dilucidar es si la generación de especies reactivas de oxígeno mediada por las diferentes isoformas de esta enzima en los distintos tipos celulares puede ser utilizada como herramienta terapéutica. Por ejemplo, en el caso del endotelio y la adventicia la producción de EROS está principalmente mediada por la activación de NOX-2, mientras que NOX-1 y/o 4 parecen ser las principales isoformas generadoras de O2- en las células de músculo liso vascular16.
La AII está directamente implicada en la formación de especies reactivas de oxígeno y en la expresión de las distintas subunidades de la NADPH oxidasa en células endoteliales y de músculo liso vascular17,18.
En la última década, diversos estudios han demostrado la importancia de las especies reactivas de oxígeno producidas por la NADPH oxidasa en las cascadas de señalización de la AII, así como la participación de estas EROS en las diferentes patologías en las que la AII es un componente central19–21. Así, las EROS derivadas de la NADPH oxidasa juegan un papel clave en el desarrollo de la hipertensión inducida por AII22–24, debido a que se encuentran implicadas en la mayoría de los procesos que subyacen al remodelado cardiovascular hipertensivo, tales como la disfunción endotelial25, la inflamación, la hipertrofia, la apoptosis, la migración, la fibrosis y la angiogénesis26.
Si consideramos que la AII incrementa la producción de radicales libres, que es uno de los procesos claves en el desarrollo aterosclerótico, se podría considerar que los fármacos que bloquean el sistema renina-angiotensina podrían actuar como antioxidantes previniendo el daño vascular. En este sentido se ha observado recientemente que la administración de un inhibidor de la enzima de conversión de la angiotensina II (IECA) en conejos hiperlipidémicos reduce la actividad de la NADPH oxidasa, eleva los niveles vasculares de tetrahidrobiopterina (limitando el desacoplamiento de la óxido-nítrico-sintasa endotelial) y aumenta la biodisponibilidad del NO, reduciéndose además la formación de placa aterosclerótica27. Asimismo se ha demostrado la mejor funcionalidad de la NOSe tras la administración de un antagonista de los receptores AT1 de angiotensina II (ARA II) en ratas diabéticas28. Además, en un modelo de hiperlipidemia experimental se ha demostrado que la combinación de ambas clases de fármacos (IECA y ARA II) puede poseer efectos protectores aditivos aumentando la disponibilidad de NO y reduciendo en mayor medida el tamaño de la placa aterosclerótica29.
Angiotensina II y disfunción endotelialLas fases más precoces de la arteriosclerosis se caracterizan por la disfunción endotelial, que precipitará la adhesión e infiltración progresiva de los monocitos circulantes que se diferenciarán a macrófagos en el subendotelio. En este contexto se ha demostrado que la AII regula el crecimiento y la migración de las células musculares y fibroblastos, la apoptosis de las células endoteliales y la diferenciación de monocitos a macrófagos30.
La AII ejerce su acción vasoconstrictora a través de los receptores AT1 de las células del músculo liso vascular, pero además induce la liberación de tromboxano A2 y endotelina-1 en células de músculo liso y endoteliales31,32, factores que aumentan las resistencias periféricas y por lo tanto la presión arterial. Asimismo el estrés oxidativo que la AII genera en la pared vascular contribuye a la disfunción endotelial, ya que el incremento en aniones O2- disminuye la biodisponibilidad del NO33.
En este sentido, nuestro grupo demostró que el tratamiento con un IECA o con un ARA II en conejos alimentados con una dieta rica en colesterol no sólo mejoraba la relajación a acetilcolina, sino que también reducía la vasoconstricción dependiente del endotelio34,35. En animales hipercolesterolémicos la alteración en la función endotelial se asocia con un aumento de los receptores AT1 y de la actividad de la NADPH oxidasa.
La posibilidad de un efecto beneficioso del bloqueo del sistema renina-angiotensina en las etapas iniciales del proceso aterosclerótico —es decir, en la disfunción endotelial— queda patente en diversos estudios clínicos. Las primeras evidencias se obtuvieron en el estudio TREND, en el que se observó que el tratamiento con quinapril mejoró la disfunción endotelial en pacientes con enfermedad arterial coronaria36. Resultados similares se obtuvieron con valsartán, que mejoró la producción y la liberación basal de NO en pacientes hipertensos en comparación con los tratados con diuréticos, a pesar del descenso similar en los niveles de presión arterial37. Es necesario tener en cuenta que mientras los ARA II ejercen sus acciones sobre la función endotelial de manera similar (a través de los receptores AT1), en el caso de los IECA debemos considerar la existencia de ECA plasmática, que regula la presión arterial, y de ECA tisular, implicada en la regulación de la inflamación tisular, la fibrosis y la hipertrofia38. En el estudio BANFF, por ejemplo, la administración de enalapril, un IECA con baja actividad a nivel tisular, no fue capaz de mejorar la función endotelial en pacientes con enfermedad coronaria39. Más recientemente, el estudio TRENDY comparó un IECA —el ramipril— con un ARA II —el telmisartán— en términos de mejora de la función endotelial40; no se encontraron diferencias entre los dos grupos, aunque la mejora observada con el ARA II fue algo más eficiente.
Angiotensina II e inflamaciónAl principio de los noventa, Dzau y Braunwald41 propusieron el concepto del continuo cardiovascular en humanos, por el que la enfermedad cardiovascular puede considerarse una cascada fisiopatológica inducida por la presencia de factores de riesgo como la hipertensión, la hipercolesterolemia, la diabetes y el tabaquismo. Estas condiciones pueden producir estados bien definidos, como la disfunción endotelial, la aterosclerosis y el daño en órganos diana, seguidos en último lugar por los síndromes clínicos (insuficiencia cardiaca, infarto de miocardio o enfermedad renal). Existen evidencias suficientes para confirmar el papel clave del sistema renina-angiotensina en el proceso inflamatorio y en todos y cada uno de los estados del continuo, lo que sugiere la importancia de su bloqueo con el fin de prevenir episodios cardiovasculares42.
En las últimas décadas se ha sugerido que el bloqueo del sistema renina-angiotensina ejerce potentes efectos antiateroscleróticos, no sólo por su efecto antihipertensivo sino a través de sus propiedades antiinflamatorias, antiproliferativas y antioxidantes42. Así, en diversos modelos animales con tendencia a desarrollar aterosclerosis se ha demostrado que la administración de IECA reduce las lesiones ateroscleróticas independientemente de los niveles de presión arterial43–48.
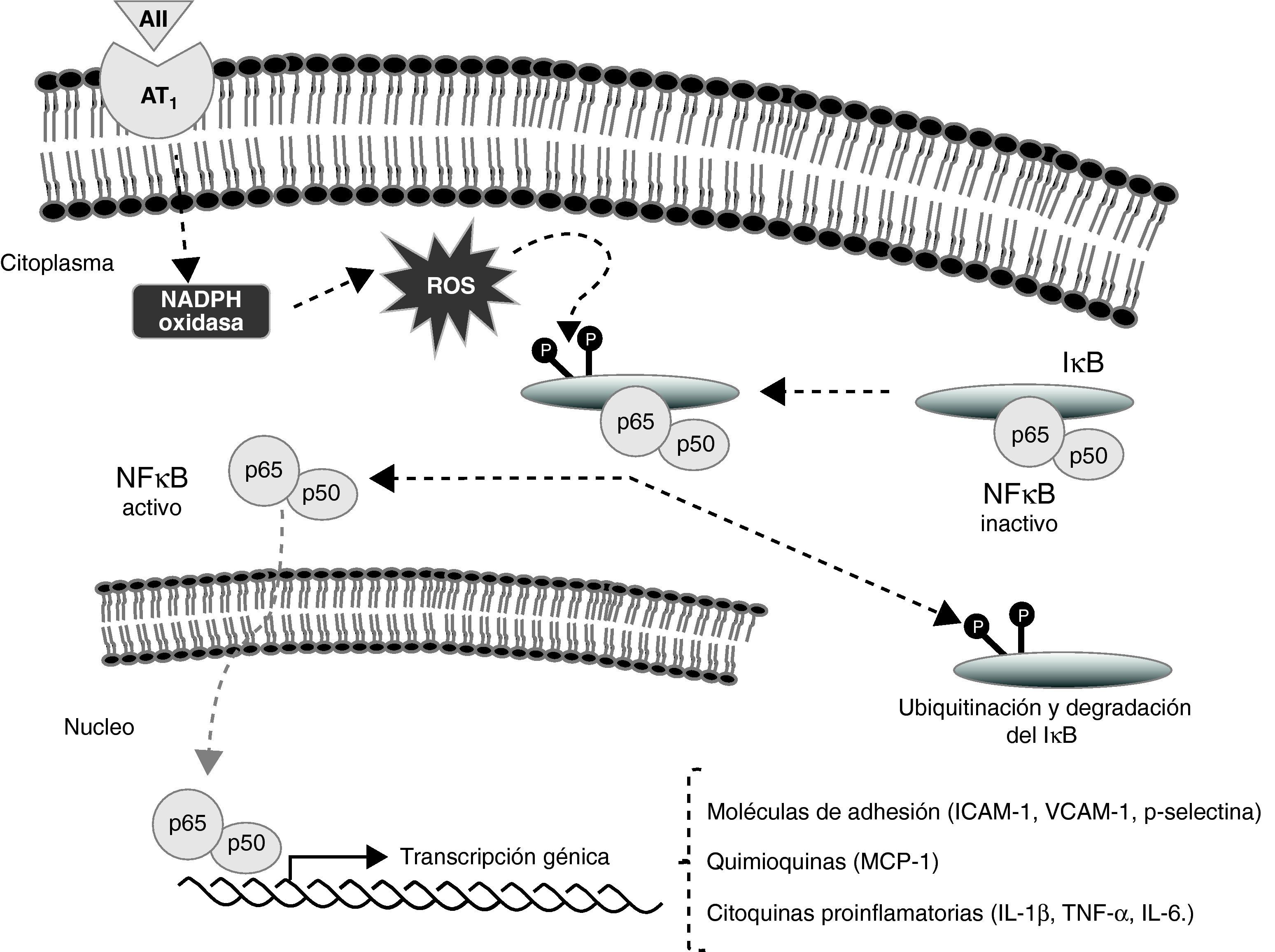
Muchos autores definen la progresión de la lesión aterosclerótica como un proceso inflamatorio continuo en la pared vascular49,50. En los últimos años, el papel del factor de transcripción NFκB en el proceso inflamatorio ha sido objeto de un profundo estudio, llegando a proponerse como un sistema de señalización que unifica el proceso inflamatorio en la aterosclerosis51. La AII, a través principalmente de receptores AT1, es capaz de activar el NFκB y provocar su translocación al núcleo en monocitos51, células del músculo liso vascular52 y células endoteliales53.
La activación del NFκB por la AII es mediada en parte por la activación de la NADPH oxidasa y el aumento consiguiente de radicales libres. Estos activan la ruta de señalización del NFκB de forma que su inhibidor el IκB se fosforila, permitiendo la liberación y la translocación al núcleo de este factor de transcripción. Una vez en el núcleo el NFκB tiene múltiples efectos en los diferentes tipos celulares de la pared vascular (fig. 3) e induce la expresión de moléculas de adhesión, tales como ICAM-1, VCAM-1 y p-selectina en células endoteliales y musculares lisas53–55, promoviendo la adherencia, la migración y la acumulación de monocitos y linfocitos T en el espacio subendotelial56. Estudios experimentales han mostrado un incremento en la expresión de VCAM-1 en aortas de animales perfundidos con AII55, así como un aumento de ICAM-1 y p-selectina en células endoteliales estimuladas con este péptido53,55. Por lo tanto, en lesiones ateroscleróticas la AII parece promover la adhesión de leucocitos a células endoteliales y de músculo liso vascular mediante la regulación en la expresión de moléculas de adhesión de la superficie celular57.

La angiotensina II participa en la adhesión de monocitos y en el proceso inflamatorio que subyace al desarrollo de la aterosclerosis al aumentar la producción de moléculas de adhesión, quimiocinas y citocinas proinflamatorias. Este efecto está mediado por la estimulación de factores de transcripción como el NFκB.
Asimismo, la AII parece ser capaz de incrementar la expresión de quimiocinas como el MCP-1 por parte de los macrófagos y las células musculares lisas, reclutando monocitos y linfocitos T al interior de la pared vascular58. En un modelo animal de aterosclerosis el tratamiento con un IECA redujo la expresión vascular de quimiocinas como el MCP-1 o la IL-859,60. Existen también estudios clínicos que demuestran una disminución de los valores plasmáticos de MCP-1 tras la administración de un IECA en pacientes que habían padecido un infarto de miocardio61.
En el proceso inflamatorio de las lesiones ateroscleróticas están implicadas diversas citocinas proinflamatorias, tales como la IL-1β, el TNF-α y la IL-6. Esta última se encuentra además implicada en la estimulación de enzimas que degradan la matriz extracelular (metaloproteinasas) y en la migración de células de músculo liso62, contribuyendo en último término a la inestabilidad de la placa. Estudios experimentales demuestran que la AII es capaz de incrementar la expresión de IL-6 en macrófagos y células de músculo liso vascular63–65. Asimismo, el estudio de arterias coronarias de pacientes que habían padecido un infarto de miocardio revela la colocalización de la IL-6 con el receptor AT1 de AII, la ECA y la AII en macrófagos cercanos a sitios susceptibles de rotura de la placa63.
Numerosos estudios clínicos han demostrado la eficacia de los IECA y los ARA II en la reducción de los niveles séricos de parámetros inflamatorios (TNF-α, IL-6, IL-10, IL-1β, PCR), moléculas de adhesión (ICAM-1, VCAM-1, e-selectina) y quimiocinas en una gran variedad de enfermedades (enfermedad arterial coronaria, insuficiencia cardiaca, hipertensión esencial, infarto de miocardio, diabetes y artritis reumatoide)66–75.
Recientemente se ha descrito la vía de señalización mediante la cual la AII es capaz de incrementar la expresión de PCR en macrófagos76. Además se ha descrito que esta proteína es capaz de aumentar la expresión de receptores AT1 de AII en células de músculo liso vascular77, lo que potenciaría la acción del péptido en cuestión.
En 1997 se aprobó el uso de un inhibidor directo de la renina, el aliskiren, adecuado en el tratamiento de la hipertensión en los casos en los que no hubiese fallo renal. La inhibición de la renina bloquea la síntesis de AI y consecuentemente de AII. Estudios muy recientes sugieren un papel directo de los inhibidores de la renina en la disminución de la inflamación asociada al proceso aterosclerótico78–80, previniendo la disfunción endotelial y la aterosclerosis en conejos hiperlipidémicos80, ratones deficientes en ApoE78 o deficientes en el receptor de LDL81.
Angiotensina II y lesión ateroscleróticaAunque existen algunos estudios iniciales en los que se cuestionaba la capacidad de los bloqueadores de los receptores de AII para reducir el tamaño de la lesión aterosclerótica82,83, en la actualidad se considera que tanto este grupo de fármacos como los IECA son eficaces en la reducción de la lesión en modelos experimentales de arteriosclerosis. Esta reducción en el tamaño de la lesión se produce como consecuencia de una disminución de todos sus componentes, es decir, un menor contenido en células espumosas, células musculares, contenido lipídico y matriz extracelular. En algunos estudios esta mejora se asoció a una reducción moderada de los niveles de presión arterial, lo que sugería que esta mejora podría ser mediada, al menos en parte, por un menor estrés hemodinámico sobre la pared vascular84. Sin embargo, diversos estudios han demostrado que estos fármacos pueden reducir el tamaño de la lesión aterosclerótica sin modificar los niveles de presión arterial ni de colesterol plasmático35,85. Asimismo, hemos observado en conejos ateroscleróticos que la reducción de la lesión producida por la administración bien de un IECA o bien de un ARA II se correlaciona negativamente con la relajación máxima a acetilcolina, sugiriendo que la reducción en el engrosamiento de la íntima puede estar implicada en la mejora de la función endotelial observada en estos animales al reducir la barrera física que separa el NO de las células musculares34,35.
Existen estudios clínicos que evidencian que los inhibidores del sistema renina-angiotensina también tienen efecto antiaterosclerótico. En el estudio SECURE86, el ramipril en forma dependiente de la dosis empleada demostró una reducción en la progresión de la aterosclerosis. En cuanto a los ARA II, existen ensayos clínicos (estudios VIOS87 y MORE88) que también evidencian mejorías en el grosor íntima-media arterial89. En concreto, en el estudio MORE se demostró por primera vez que la administración de un ARA II, en concreto el olmesartán, fue capaz de reducir significativamente el volumen de grandes placas ateroscleróticas, en comparación con el atenolol, en pacientes hipertensos. En un subanálisis con pacientes cuyos valores iniciales mostraban placas por encima de la media, 2 años de tratamiento con olmesartán redujeron el volumen de la placa en un 8,9%, comparada con un incremento del 2,4% con atenolol. En placas incluso mayores, el volumen de la placa (VP) se redujo un 12,8% y aumentó un 2,1%, respectivamente, en los grupos de olmesartán y atenolol. Estos efectos regresivos del VP, mostrados por el olmesartán sobre placas grandes, fueron evidentes a partir de las 28 semanas.
El estudio clínico más reciente al respecto es el J-ELAN, en el que se compara el efecto de un ARA II (losartán) frente a un bloqueador de los canales del calcio (amlodipino) en pacientes con hipertensión leve-moderada. En él se demuestra que el ARA II ante una reducción de presión similar es más efectivo en la reducción de la progresión de la lesión aterosclerótica que el amlodipino, mejorando además la función diastólica del ventrículo izquierdo90. Sin embargo, el estudio MITEC91, realizado en pacientes diabéticos con hipertensión leve-moderada, mostró una reducción similar en el grosor íntima-media carotídeo entre otro ARA II (candesartán cilexetil) y el amlodipino. De la misma manera, en el estudio AAA92 no se observaron cambios en el grosor íntima-media carotídeo en pacientes diabéticos e hipertensos tras la administración de diferentes ARA II (losartán, candesartán, valsartán y telmisartán), mientras que el amlodipino sí fue capaz de producir un descenso significativo de este parámetro. No obstante, tras el análisis detallado del estudio LIFE se puede concluir que el losartán puede tener una serie de propiedades independientes de la disminución de la presión arterial, que pueden estar asociadas a una disminución en la vulnerabilidad de la placa93,94.
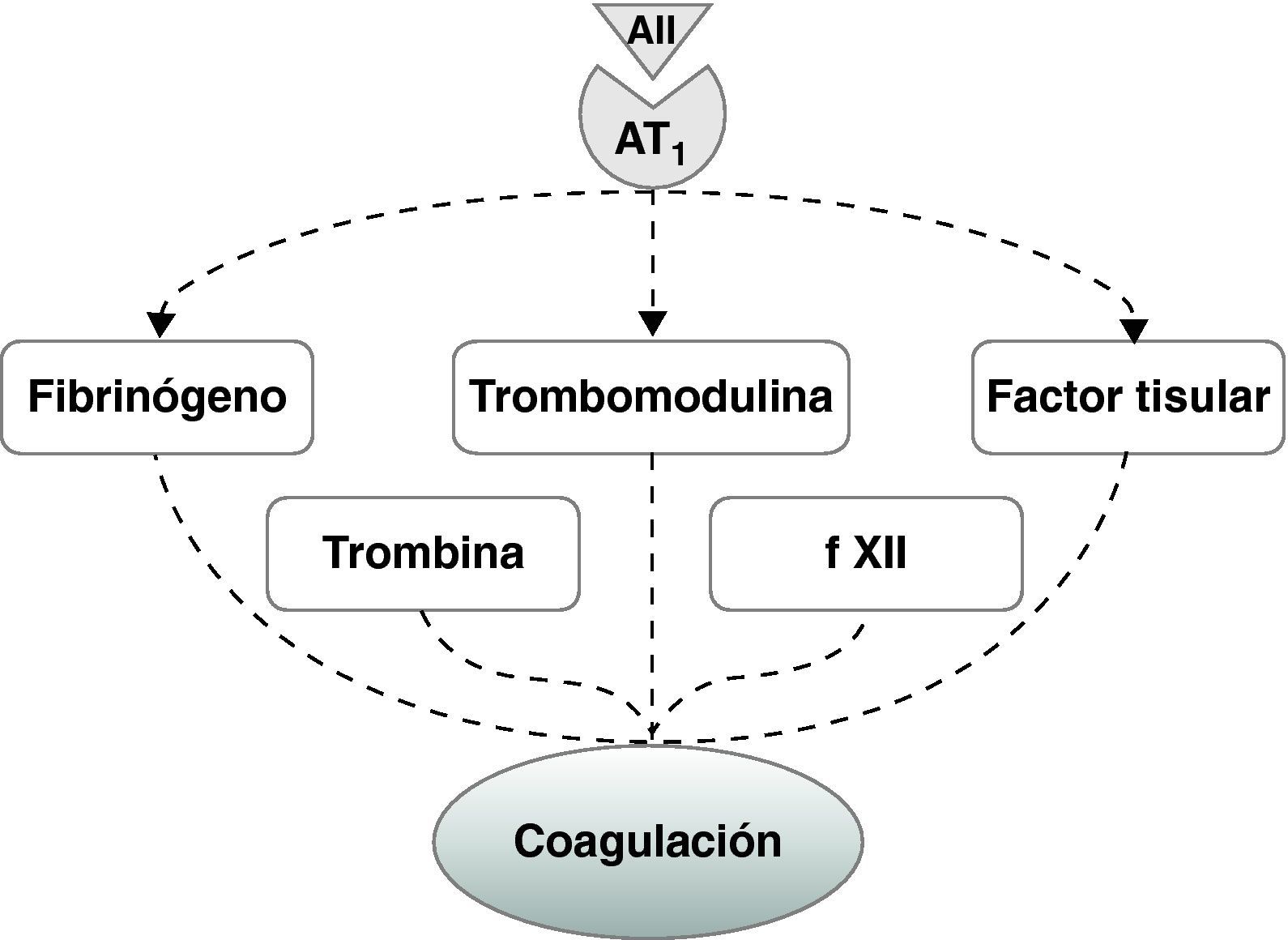
Angiotensina II, coagulación y fibrinólisisNumerosos estudios experimentales indican que el bloqueo del sistema renina-angiotensina podría tener efectos beneficiosos sobre la agregación plaquetaria, la coagulación y la fibrinólisis. A grandes rasgos, los IECA parecen mejorar el balance fibrinolítico disminuyendo los niveles de PAI-1 plasmáticos, y los ARA II actúan como antiagregantes plaquetarios95,96, aunque también existen estudios que demuestran actividad antifibrinolítica de estos últimos mediante la reducción en la expresión de PAI-197–100.
En concreto, se ha demostrado que en plaquetas humanas aisladas los ARA II reducen la liberación de tromboxano A2 y de calcio inducida por la trombina101. Asimismo, se ha demostrado que los ARA II reducen la activación plaquetaria in vitro mediante el bloqueo del receptor de tromboxano A2 y su vía de señalización, y reducen la agregación plaquetaria producida por ADP y trombina101. En relación con el sistema de coagulación, también se ha demostrado que el bloqueo del sistema renina-angiotensina tiene un efecto beneficioso sobre la coagulación. El tratamiento durante 6 meses con un ARA II en pacientes hipertensos se asoció con una disminución de las concentraciones plasmáticas de trombomodulina, factor XII, fibrinógeno y factor tisular102 (fig. 4). El efecto beneficioso sobre el balance fibrinolítico se observa claramente en estudios experimentales. En este sentido, en conejos alimentados con una dieta enriquecida con un 1% de colesterol hemos observado que la administración de un ARA II fue capaz de prevenir el aumento de la actividad de PAI-1 como la disminución de la actividad de t-PA producida por la hipercolesterolemia103. Este efecto se asoció con una mejora de la función endotelial y una reducción de la lesión aterosclerótica, lo que sugiere que el efecto beneficioso de estos fármacos sobre la fibrinólisis parece estar relacionado con la mejora concomitante de la función endotelial y la reducción de la progresión aterosclerótica. Este efecto fue independiente de los valores de presión arterial y de colesterol plasmático, lo que sugiere que no se debe a efectos hemodinámicos o sobre el perfil lipídico ejercidos por estos fármacos103. De manera similar, los estudios clínicos han demostrado también la eficacia de los ARA II sobre los factores que regulan el equilibrio fibrinolítico. En pacientes hipertensos y con insuficiencia cardíaca la administración de un ARA II redujo los valores de PAI-1 e incrementó los de t-PA, mejorando de esta manera el alterado balance fibrinolítico98,104,105.

En resumen, todos estos datos indican que la AII afecta, mediante los receptores AT1, la regulación de diversas funciones relacionadas con el desarrollo y progresión de la lesión aterosclerótica. Por este motivo una sobreactivación del sistema renina-angiotensina puede aumentar el riesgo de episodios cardíacos incluso independientemente de su acción sobre los valores de presión arterial. Todo esto justifica que la AII sea considerada como uno de los principales factores vasoactivos implicados en la patología cardiovascular y que la administración de fármacos que modulan este sistema ejerza un efecto beneficioso sobre el desarrollo aterosclerótico, previniendo las alteraciones trombóticas asociadas a élcc.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.