




El colesterol es necesario para la proliferación y la correcta progresión del ciclo celular. La inhibición sostenida de la biosíntesis de colesterol inhibe la citocinesis y da lugar a la aparición de células poliploides, efectos que pueden implicar la participación de las vías de estrés.
ObjetivoEstudiar el efecto de la inhibición terminal de la biosíntesis de colesterol sobre la vía de p38 MAPK y su papel en la progresión del ciclo celular.
MetodologíaLa inhibición de la biosíntesis de colesterol en las células promielocíticas humanas HL-60 se llevó a cabo con SKF 104976. En determinados casos, junto a este inhibidor, se inhibieron las vías de p38 MAPK y ERK1/2 empleando SB 203580 y PD 98059, respectivamente. El ciclo celular se estudió por marcaje con yoduro de propidio e incorporación de bromodesoxiuridina y análisis por citometría de flujo, y la expresión de proteínas se analizó por Western blot.
ResultadosLa inhibición de la biosíntesis de colesterol produjo la acumulación de células en G2/M y una activación transitoria de p38 MAPK, efectos que fueron revertidos por las lipoproteínas de baja densidad (LDL), demostrando que se debían a la deficiencia de colesterol. En aquellas condiciones, la adición de SB 203580 aceleró la replicación del ADN acompañada de un aumento de la poliploidía. Contrariamente, la adición de PD 98059 inhibió la síntesis de ADN. Ni la inhibición de p38 MAPK ni la de ERK impidieron la división de las células previamente tratadas con SKF 104976 tras suplementarlas con LDL, ni de las células previamente sincronizadas en prometafase con nocodazol.
ConclusionesLa inhibición de la biosíntesis de colesterol activa la vía de p38 MAPK a fin de impedir la poliploidía pero no tiene efecto sobre la culminación de la mitosis.
Cells require cholesterol for proliferation and correct progression of the cell cycle. In the absence of cholesterol, the cells fail to undergo cytokinesis and polynucleated cells are generated. These effects could be mediated by stress signal transduction pathways.
ObjectiveTo study the effects of cholesterol biosynthesis inhibition on the activity of p38 MAPK and to evaluate the role of this pathway on cell cycle progression.
MethodologyHuman leukemia cells (HL-60) were incubated in the presence of SKF 104976, an inhibitor of cholesterol biosynthesis. In some cases inhibitors of p38 MAPK and ERK1/2, namely SB 203580 and PD 98059, were added to the medium. Cell cycle progression was studied by flow citometry, both DNA content and bromodeoxyuridine incorporation into DNA, and protein expression of p38 MAPK was analyzed by western blot.
ResultsInhibition of cholesterol biosynthesis led to accumulation of cells in G2/M and a transient activation of p38 MAPK. These effects were reversed by supplementing the medium with LDL. The addition of SB 203580 accelerated DNA replication, which was accompanied by an increase of polyploidy. By contrast, the addition of PD 98059 inhibited DNA synthesis. Lastly, neither the inhibition of p38 MAPK nor ERK affected the division of cells treated with the cholesterol biosynthesis inhibitor following LDL provision, or mitosis completion from methaphase of cells previously synchronized with nocodazole.
ConclusionsCholesterol deficiency induces p38 MAPK pathway activation in order to prevent polyploidy, but has no effect on mitosis completion.
El colesterol es necesario para la proliferación celular. En este sentido, el colesterol no sólo es un componente integral de la membrana celular sino que también parece ejercer acciones reguladoras que permiten la correcta progresión del ciclo celular. En la línea promielocítica HL-60, hemos demostrado que la deficiencia de colesterol producida por inhibición de la biosíntesis de colesterol en células incubadas en un medio libre de colesterol, produce una serie de efectos que, en último término, impiden la división celular. Inicialmente se observa una retención de las células en G2/M, con inhibición de la citocinesis, que finalmente conduce a la formación de células poliploides por un mecanismo de rerreplicación1. Todos estos efectos se evitan y revierten, al menos parcialmente, añadiendo colesterol al medio de cultivo, lo que demuestra que las acciones que ejerce el colesterol en esas etapas del ciclo celular son específicas2–4.
La progresión del ciclo celular se produce de una forma ordenada, lo que está mediado por la activación puntual de distintos complejos ciclina/Cdk y su desactivación posterior. Concretamente, la transición de G2 a mitosis está controlada por el complejo ciclina B1/Cdk1, cuya actividad está gobernada por expresión de la ciclina B1, su localización subcelular y la fosforilación de la Cdk1. Pues bien, hemos demostrado que la parada en G2/M producida por la deficiencia experimental de colesterol coincide con la disminución de la actividad de ciclina B1/Cdk1 y que la reposición de colesterol estimula la transcripción del gen de la ciclina B1 y, consiguientemente, la actividad del complejo3,4.
Las células que superan ese punto de control de G2, transcurren por mitosis hasta completar la cariocinesis, pero ven impedida la citocinesis1. El requerimiento de colesterol para que se produzca la abscisión celular ha sido confirmado por otros investigadores5. Estos autores observaron que en los momentos previos a la citocinesis, el colesterol de la membrana es reclutado hacia el anillo mitótico y que la extracción selectiva de colesterol mediante metil-beta-ciclodextina impide la citocinesis5.
Este papel del colesterol en la mitosis coincide con la estimulación de la biosíntesis de este lípido precisamente en esta etapa, como recientemente demostraron Bengoechea-Alonso et al6. Es interesante que la ciclina B1/Cdk1 es capaz de fosforilar SREBP en una zona próxima al dominio "phosphodegron", evitando su degradación por el proteasoma7, lo que explicaría la estimulación de la transcripción de los genes relacionados con la biosíntesis de colesterol y el receptor de lipoproteínas de baja densidad (LDL) precisamente en mitosis.
La deficiencia de colesterol se puede considerar como un estrés ante el cual la célula puede responder activando algunas de las denominadas vías de estrés, entre las que se encuentra la de p38 MAPK (p38 mitogen-activated protein kinase). Esta vía se activa en respuesta a citocinas proinflamatorias y distintos tipos de estrés, como la radiación UV y los choques térmico y osmótico8–13. Por lo general, dichos estímulos son recogidos por las cinasas MKK3/6 que fosforilan a p38 en sendos residuos Thr y Tyr activándola. La activación de esta vía conduce a la inhibición de la proliferación celular, como se ha demostrado repetidamente. Por ejemplo, la activación de p38 en G1 inhibe la expresión de la ciclina D1, lo cual impide la transición a S14,15. En la transición de G2 a M, la activación de p38 MAPK por daño en el ADN conduce a la degradación de Cdc25C, lo cual impide la transición a M por falta de activación del complejo ciclina B1/Cdk116. Por lo tanto, p38 MAPK actúa como una proteína reguladora que impide la proliferación celular en respuesta al daño celular12 y, de hecho, la inhibición de la vía de p38 MAPK resulta en la estimulación de distintos genes relacionados con la proliferación celular y en un aumento del índice mitótico en diferentes líneas celulares11,17,18. p38 MAPK se considera actualmente como un importante gen supresor de tumores, como p5310,12. Recientemente se ha demostrado que p38 MAPK participa activamente en la regulación fisiológica de la transición a mitosis en ausencia de estrés19.
Otros autores han establecido una conexión entre el metabolismo de lípidos y la vía de p38 MAPK, en el sentido de que la activación de esta vía produce una inhibición de la expresión del receptor de LDL20 mientras que la inhibición de p38 MAPK induce la expresión de dicho receptor9.
En el presente trabajo se ha estudiado el papel de p38 MAPK en los efectos producidos por la inhibición terminal de la biosíntesis de colesterol en la progresión del ciclo celular. Se ha observado que la inhibición de la biosíntesis de colesterol produce la activación de p38 MAPK y que, en esas condiciones, la inhibición de ésta conduce a la poliploidía. Estos resultados sugieren que en respuesta a la deficiencia de colesterol, la célula responde activando p38 MAPK, tratando de frenar la replicación del ADN.
MétodosCultivos celularesLas células de la línea promielocítica humana HL-60 (ATCC CCL 240) se mantuvieron en un medio libre de colesterol DCCM-1 (Biological Industries), que contenía penicilina 100 U/ml, estreptomicina 100 U/ml y gentamicina 100μg/ml (Gibco, BRL). Para la inhibición de la biosíntesis de colesterol, las células se incubaron en presencia de SKF 104976 1,5μM (SmithKline Beecham), un inhibidor de la lanosterol 14α-desmetilasa3,21, disuelto en DMSO (concentración final en el medio, 0,04%). En determinados casos, el medio se suplemento con colesterol libre disuelto en etanol (concentración final en el medio, 0,4%) o bien LDL humanas aisladas por ultracentrifugación. Para la inhibición de las vías de p38 MAPK y de ERK, las células se incubaron en presencia de los inhibidores específicos SB 203580 20μM y PD 98059 50μM, respectivamente. Para sincronizar las células en prometafase, se añadió al medio nocodazol 0,05μg/ml, un inhibidor de la formación de los microtúbulos.
Análisis del ciclo celularLas células se cultivaron a una densidad de 3 × 105 células/ml y tras los diferentes tratamientos experimentales se recogieron por centrifugación y se fijaron en etanol al 70% durante 30min a −20°C. Después del proceso de fijación se lavaron con PBS y se incubaron durante 1h a 37°C en una solución de ribonucleasa A 100μg/ml y yoduro de propidio 50μg/ml. Posteriormente se analizaron mediante citometría de flujo (FACScalibur, Becton-Dickinson), excluyéndose los dobletes utilizando la representación de área (FL2-A) frente a la anchura (FL2-W).
Análisis de la fase S activa del ciclo celular por incorporación de bromodesoxiuridinaTras los diferentes tratamientos experimentales se recogieron 2–4 × 106 células y se incubaron durante 1h en presencia de bromodesoxiuridina (BrdU) 100μM (Sigma Aldrich) a 37°C. A continuación las células se lavaron con PBS y se fijaron en etanol al 70% durante 30min a −20°C. Una vez fijadas se incubaron durante 20min en HCl 2N a temperatura ambiente y transcurrido ese tiempo se lavaron 3 veces con PBS. Después las células se resuspendieron en 1ml de tampón tetraborato de disodio (Na2B4O7 × 10H2O 0,1M pH 8,4), se centrifugaron, se lavaron una vez más con PBS y se resuspendieron en una solución de bloqueo compuesta por 500μl de PBS, 0,5% Tween 20 (Sigma Aldrich), 1% NGS (Normal Goat Serum, Vector) durante 15min a temperatura ambiente. Finalizado el proceso de bloqueo las células se centrifugaron y resuspendieron en la misma solución, a la que se añadieron 20μl de anticuerpo anti-BrdU-FITC (Becton Dickinson), incubándolas durante 2h en oscuridad y a temperatura ambiente. Por último, las células se lavaron 2 veces con PBS, 0,5% Tween 20 (Sigma Aldrich) y se tiñeron con yoduro de propidio 50μg/ ml en una solución que contenía 100μg/ml de ribonucleasa A durante 1h a 37°C. Finalizado este proceso se analizaron en el citómetro de flujo (FACScalibur, Becton-Dickinson).
Electroforesis e inmunodetección (Western blot)Finalizados los diferentes tratamientos, se recogieron 4–8 × 106 células y se lisaron con un tampón Tris–HCl 50mM pH 8, NaCl 120mM, Nonidet P40 al 0,5%, NaF 100mM, ortovanadato 1mM, PMSF 40μg/ml, aprotinina 40μg/ml y leupeptina 40μg/ml. Cantidades iguales de proteína analizada mediante el método de Bradford22, se sometieron a electroforesis en geles de poliacrilamida en condiciones reductoras y desnaturalizantes (SDS-PAGE) y, posteriormente, se realizó la transferencia a membranas de PVDF (Hybond-P, Amersham). Para la detección de la proteína p38 MAPK, total y fosforilada (Thr189/Tyr182), se utilizaron los correspondientes anticuerpos policlonales específicos (Cell Signaling). Los lavados entre el anticuerpo primario y el secundario se realizaron con leche en polvo al 0,25% en TBS-0,05% Tween 20. La señal se detectó mediante quimiobioluminiscencia (ECL kit RPN2108, Amersham Pharmacia Biotech).
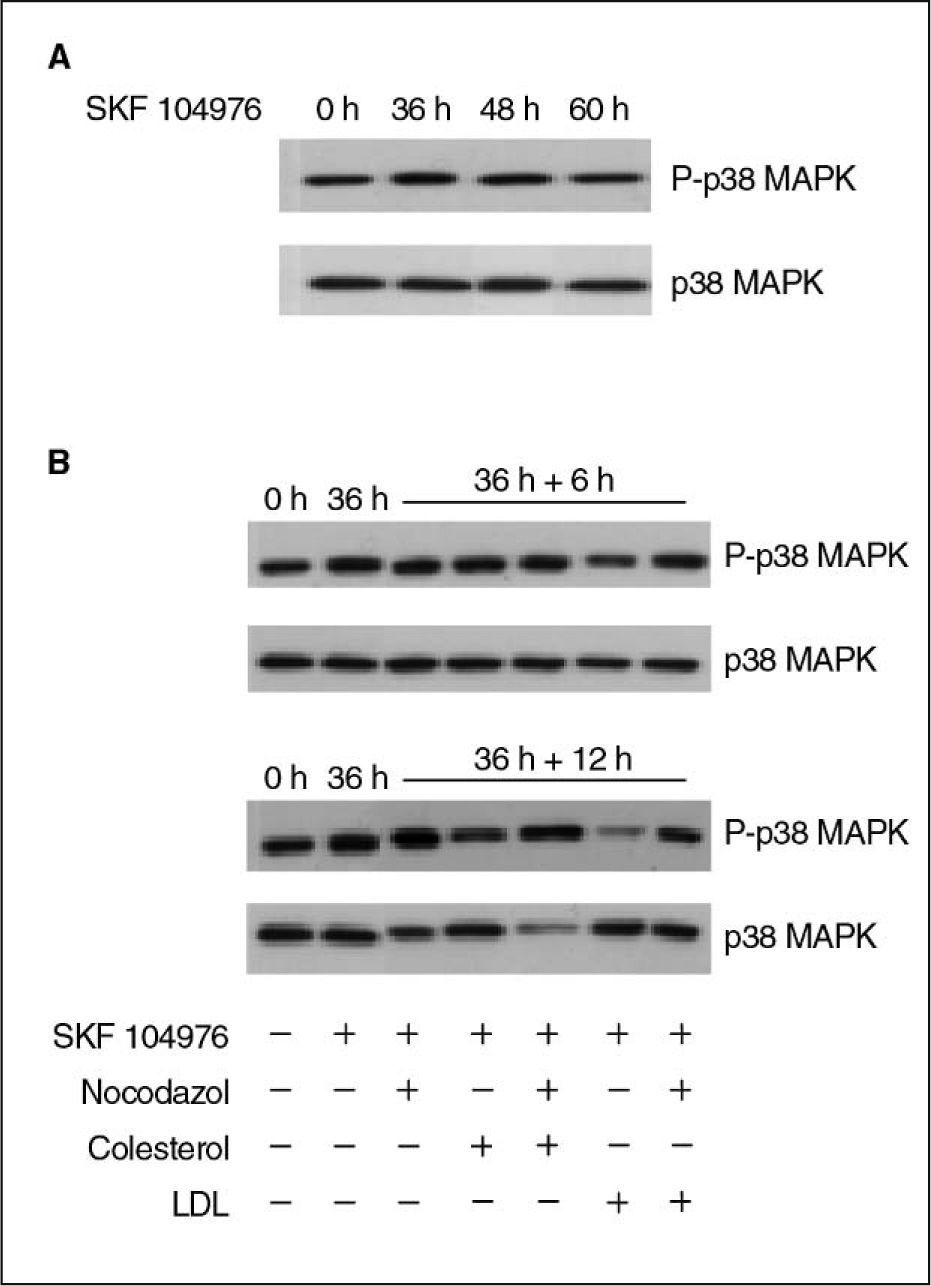
ResultadosLa inhibición de la síntesis de colesterol con SKF 104976 produce un aumento transitorio de los valores de p38 MAPK fosforiladaEn primer lugar se estudió el efecto de la inhibición de la biosíntesis de colesterol en los valores proteicos de p38 MAPK fosforilada. Para ello las células se incubaron en presencia de SKF 104976 y dichos valores se examinaron por Western blot a diferentes tiempos. Los resultados muestran un aumento transitorio de la p38 MAPK fosforilada en las primeras 36–48h de tratamiento, para decaer posteriormente, sin cambios en la p38 total (fig. 1A).
 y p38 MAPK total en células HL-60 tratadas con SKF 104976 1,5μM. A) Efecto del tratamiento a lo largo del tiempo. B) Efectos del colesterol libre (60μg/ml) y de las LDL (20μg colesterol/ml), aisladamente o en combinación con nocodazol 0,05μg/ml; las células se incubaron en presencia de SKF 104976 1,5μM y, a las 36h, sin retirar el inhibidor, se añadieron el colesterol y el nocodazol en las distintas combinaciones, y se prolongó el cultivo durante 6 o 12h. LDL: lipoproteínas de baja densidad.")
Valores proteicos de p38 MAPK fosforilada (P-p38 MAPK) y p38 MAPK total en células HL-60 tratadas con SKF 104976 1,5μM. A) Efecto del tratamiento a lo largo del tiempo. B) Efectos del colesterol libre (60μg/ml) y de las LDL (20μg colesterol/ml), aisladamente o en combinación con nocodazol 0,05μg/ml; las células se incubaron en presencia de SKF 104976 1,5μM y, a las 36h, sin retirar el inhibidor, se añadieron el colesterol y el nocodazol en las distintas combinaciones, y se prolongó el cultivo durante 6 o 12h. LDL: lipoproteínas de baja densidad.
En trabajos anteriores se había demostrado que el tratamiento con SKF 104976 producía la parada del ciclo celular en G2/M y que la adición de colesterol en ese instante restablecía la progresión del ciclo celular2,3,23. Por tanto, nos planteamos estudiar el efecto de la adición de colesterol al medio de cultivo sobre la expresión de p38 MAPK. Para ello, las células se trataron con SKF 104976 1,5μM y a las 36h, sin retirar el inhibidor, se añadió al medio de cultivo colesterol libre o LDL, estudiándose la expresión de p38 MAPK fosforilada y total a las 6 y 12h posteriores. Este experimento se realizó tanto en ausencia como en presencia de nocodazol, en este último caso para impedir que las células progresaran más allá de prometafase. Como se puede observar en la figura 1B, la adición de colesterol produjo una disminución de la forma fosforilada de p38 MAPK, más intensa con LDL que con colesterol libre, y que se acentuaba con el tiempo. Estos efectos no se debían a cambios en la cantidad de p38 MAPK total, por cuanto ésta permaneció inalterada tras la adición de colesterol libre o LDL al medio de cultivo. El nocodazol produjo ya de por sí un incremento de p38 MAPK fosforilada, lo que concuerda con resultados anteriores de otros autores24, pero aun en ese caso la adición de colesterol al medio disminuyó su expresión.
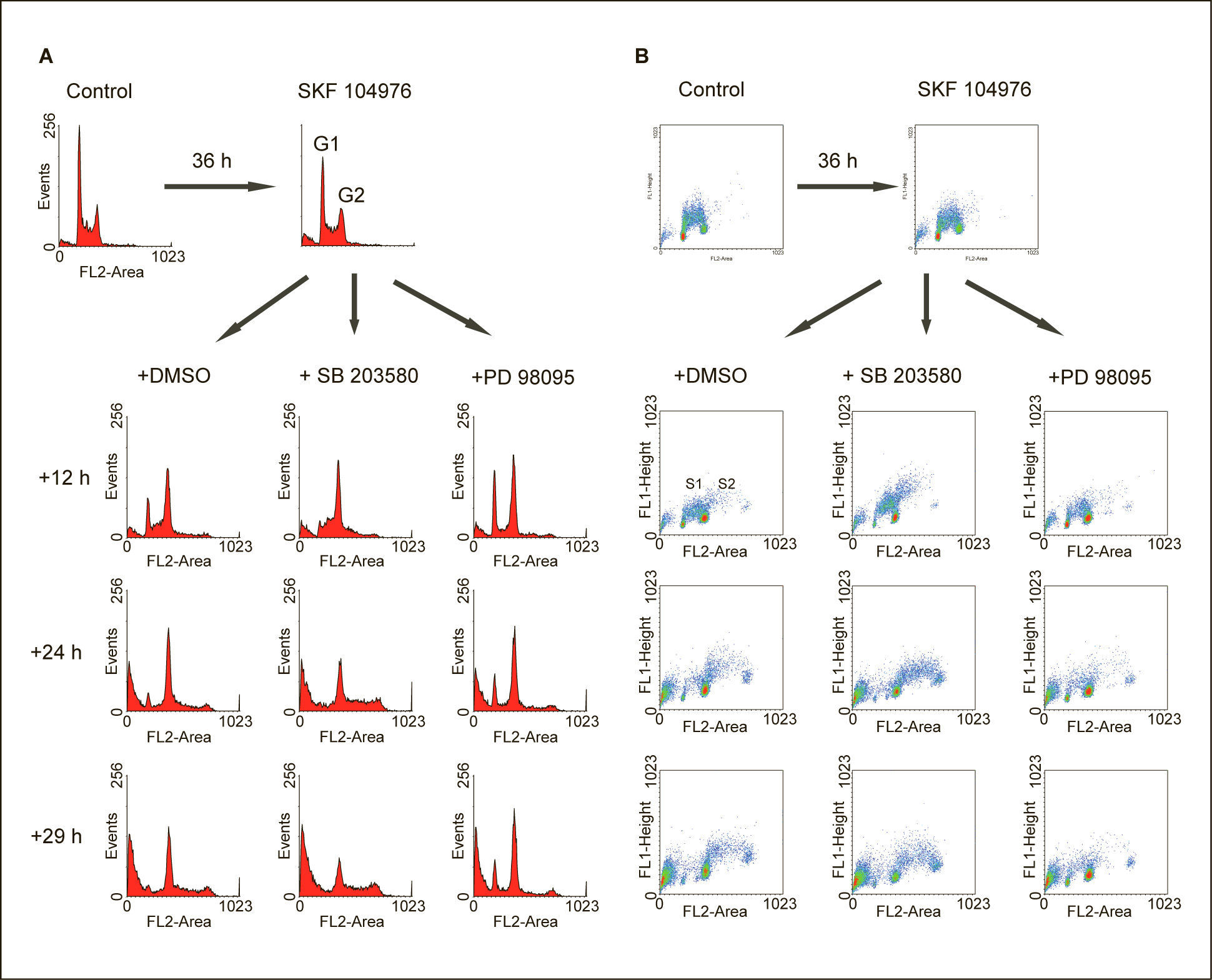
La inhibición de p38 MAPK en células deficientes de colesterol acelera la entrada en fase SLa deficiencia de colesterol conseguida a través de la inhibición de la biosíntesis de colesterol por SKF 104976 en células incubadas en un medio libre de colesterol, produjo la acumulación progresiva de células en G2/M, seguido de la aparición de células poliploides al prolongar el tiempo de incubación (fig. 2). La adición de SB 203580, un potente inhibidor de las isoformas α y β de la p38 MAPK, a células que estaban siendo tratadas con SKF 104976 produjo la práctica desaparición de las células en G1 a las 12h tras la adición de SB 203580 y el aumento de la población de células poliploides a las 24h (fig. 2A). Estos resultados sugieren que la inhibición de p38 MAPK acelera la replicación del ADN, lo cual se sustancia también en el aumento del número de células en S1 y en S2, que sintetizan activamente ADN, como se observa en los diagramas de incorporación de BrdU a las 12 y 24h tras la adición de SB 203580 en comparación con el control (fig. 2B).
 y la incorporación de BrdU al ADN (B). DMSO: dimetilsulfóxido.")
A efectos comparativos, otras células fueron tratadas con PD 98059, un inhibidor específico de ERK. Como se muestra también en la figura 2, la inhibición de la vía de ERK produjo la disminución del número de células en las fases S1 y S2 en comparación con la situación control, lo cual es compatible con una inhibición de la entrada en S tanto desde G1 como desde G2/M. Este efecto es opuesto al ejercido por SB 203580.
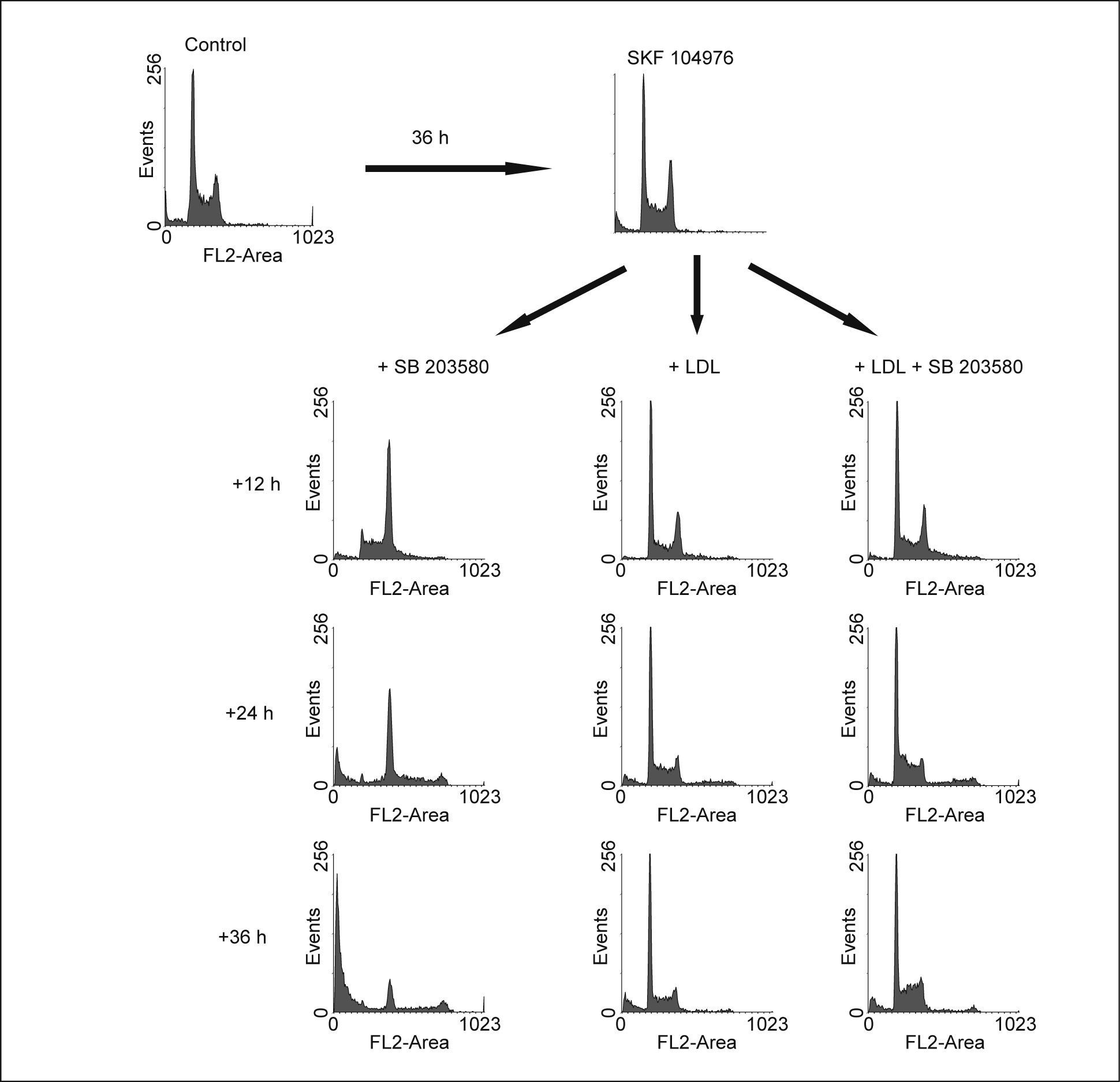
La inhibición de p38 MAPK no afecta la división celularEn esta ocasión las células fueron incubadas durante 40h en presencia de SKF 104976 y a continuación se añadieron al medio LDL, en ausencia o en presencia de SB 203580. Como se esperaba, el aporte de colesterol en forma de LDL evitó que progresara la parada del ciclo celular inducido por el SKF 104976, es más, las células anteriormente en G2/M se dividieron y aparecieron en G1, restableciéndose un ciclo celular normal a las 24h tras su adición al medio (fig. 3). La inhibición de p38 MAPK no afectó la división celular, por cuanto las células incubadas con LDL no se acumulaban en G2/M a pesar de seguir estando presente el inhibidor de la biosíntesis de colesterol. Esto significa que la inhibición de p38 MAPK no interfiere en la finalización de la mitosis. Por el contrario, SB 203580 incrementó el número de células en fase S y ligeramente también el número de células poliploides con respecto a los cultivos con LDL solas.
La inhibición de la vía de p38 MAPK en células tratadas con nocodazol no impide el restablecimiento del ciclo celular
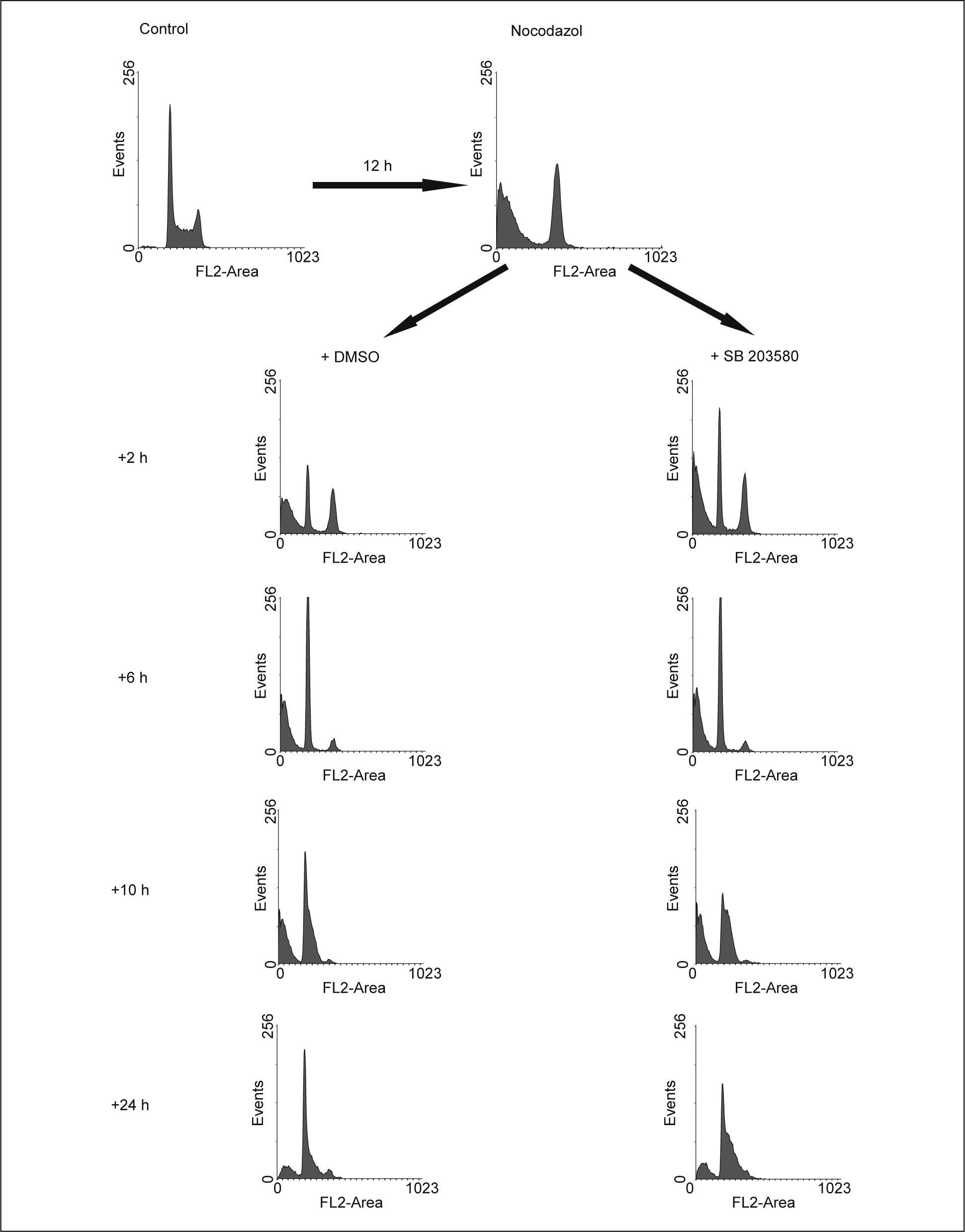
En una última aproximación para determinar si la vía de p38 MAPK participa en la finalización de la mitosis, las células fueron tratadas con nocodazol para detenerlas en prometafase, como resultado de la inhibición de la formación de los microtúbulos. Como se muestra en figura 4, el tratamiento con nocodazol durante 12h produjo la acumulación de las células en G2/M, aunque también muerte celular. Una vez retirado el nocodazol del medio, las células volvieron a ciclar libremente, restableciéndose el ciclo celular. La adición de SB 203580 una vez retirado el nocodazol, no impidió que las células anteriormente detenidas en G2/M completaran la mitosis, apareciendo en G1, no habiendo prácticamente diferencias entre los histogramas de estas 2 condiciones a las 6h después de haber retirado el nocodazol (fig. 4). Posteriormente, las células expuestas al inhibidor de p38 MAPK siguieron ciclando, pero de forma aún más acelerada que las controles, como lo muestra el incremento de la proporción de células en S a las 10 y 24h del experimento (fig. 4).
 o en presencia de SB 203580 20μM. DMSO: dimetilsulfóxido.")
Efecto de la inhibición de p38 MAPK sobre la culminación de la mitosis y la posterior entrada en fase S. Las células HL-60 fueron sincronizadas en prometafase por el tratamiento con nocodazol 0,05μg/ml, que posteriormente se retiró, dejándose proseguir en el ciclo en ausencia (DMSO) o en presencia de SB 203580 20μM. DMSO: dimetilsulfóxido.
Estos resultados son coherentes con los mostrados en las figuras anteriores y, en conjunto, indican que p38 MAPK actúa como un regulador de la entrada en la fase de síntesis de ADN, pero no parece participar en la finalización de la mitosis.
DiscusiónLas células disponen de diversas vías de señalización que les permiten responder a las señales extracelulares y a los cambios en el estado de la propia célula. Una de ellas es la vía de p38 MAPK, que se activa bajo diferentes condiciones de estrés y media distintas respuestas metabólicas y del ciclo celular11,12,25,26. En este contexto, el objetivo principal del presente trabajo ha sido determinar la participación de esta vía de señalización en los cambios en el ciclo celular provocados por la deficiencia de colesterol.
En las células leucémicas HL-60, la inhibición de la biosíntesis de colesterol con SKF 104976, un inhibidor de la lanosterol 14α-desmetilasa, produjo una acumulación inicial de las células en G2/M, ya detectable a las 36h de tratamiento, seguida de la formación de células poliploides al prolongar la incubación. Estos resultados coinciden con observaciones anteriores publicadas por nosotros1, que demostraban que el colesterol es necesario para la transición de G2 a mitosis y también para la citocinesis. Las células polinucleadas se producen, en este caso, por endorreplicación como consecuencia de la entrada en una nueva fase S (S2) de las células que no han experimentado la citocinesis1.
El ciclo celular está regido por una serie de controles (checkpoints) que garantizan que las distintas etapas del ciclo discurran de una forma ordenada y evitan que se transmitan alteraciones o defectos en la dotación celular a las células hijas. Estos controles actúan en distintas etapas y su activación detiene el avance del ciclo para permitir que se reparen las alteraciones y, si no, abortar la división celular. La poliploidía que se observa en las células deficientes de colesterol debe interpretarse, entonces, como un fallo en alguno de dichos controles, puesto que se produce la replicación del ADN en una célula con contenido genético 4n que no se ha dividido.
En el presente trabajo se ha observado que, coincidiendo con la acumulación de células en G2/M, entre las 36 y las 48h de tratamiento con el inhibidor de la biosíntesis de colesterol, se produce un incremento de la fosforilación de p38 MAPK. Esta activación era específica, puesto que se revertía al añadir colesterol al medio, lo que permite concluir que la deficiencia de colesterol se percibe por la célula como un estrés que dispara esta vía de señalización. La activación de p38 MAPK era transitoria, y coincidiendo con el descenso de los valores intracelulares de p38 MAPK fosforilada empezó a apreciarse de forma clara la formación de células poliploides. Esto sugería que p38 MAPK estaba actuando como un checkpoint, papel que ya le habían asignado otros autores10,12, pero en este caso evitando la endorreplicación en las células que, debido a su deficiencia de colesterol, no podían completar la citocinesis. Para demostrar esta posibilidad, las células se trataron con un inhibidor específico de p38 MAPK, SB 203580, que inhibe la señalización posterior27. Pues bien, este compuesto aceleró la endorreplicación, como lo indicaban el incremento del número de células que entraban en S2 desde G2/M a tiempos cortos tras su adición y la aparición posterior de células con contenido genético 8n. Por lo tanto, todo indica que p38 MAPK actúa como un control que impide que las células repliquen su ADN desfasadamente. Otros autores habían observado que la exposición de las células a distintos tipos de estrés activaba p38, retrasando la entrada en mitosis10,12. En nuestro caso, p38 media la respuesta celular a la deficiencia de colesterol. La aparición de alguna célula poliploide por efecto del tratamiento con el inhibidor de la biosíntesis de colesterol debe interpretarse, por tanto, como un fracaso parcial de este control, siendo, de hecho, como fue la activación de p38 MAPK transitoria. Las causas de que dicha activación de p38 MAPK no fuera sostenida en el tiempo se desconocen. Es conocido que las células HL-60, como muchas otras células tumorales, no expresan o expresan formas mutantes de p53, un importante gen supresor de tumores. En estas células, la respuesta al estrés, por ejemplo al daño en el ADN, descansa en la activación de p38 MAPK13,28, lo que confirma a p38 MAPK como gen supresor de tumores12. Por otro lado, se ha demostrado que la activación de p38 MAPK en respuesta a ciertos tóxicos químicos requiere p5329. Por lo tanto, esta interrelación entre p53 y p38 puede explicar el hecho de que las células carentes de p53, como las HL-60, no puedan mantener activa p38 MAPK en respuesta a la deficiencia de colesterol.
Los resultados del presente trabajo no aclaran si la interrupción de la citocinesis como consecuencia de la falta de colesterol está mediada por la activación de p38 MAPK. Su demostración exigiría la utilización de células que carecieran de p38 MAPK o de alguno de los otros componentes de esta vía. Aun así, nuestros resultados indican que no se requiere p38 MAPK para que se complete la citocinesis. Así lo evidencian los experimentos de reposición del colesterol a células tratadas con SKF 104976, donde se observa que la adición de LDL permite que las células se dividan, tanto en ausencia como en presencia del inhibidor de p38 MAPK. En el mismo sentido, SB 203580 tampoco afectó el avance por mitosis y la división de las células previamente sincronizadas en prometafase con nocodazol. Estos resultados no se contradicen con el papel que recientemente se le ha asignado a p38 MAPK en la regulación de la entrada en mitosis desde G2, donde actuaría como checkpoint13,19.
En el presente trabajo también se ha observado que la vía de ERK no es necesaria para que se complete la citocinesis, en tanto y en cuanto la inhibición de dicha proteína no impide que las células tratadas con SKF 104976 se dividan normalmente tras la adición de colesterol (LDL) al medio. En la bibliografía hay cierta controversia acerca del papel de ERK en mitosis. Mientras que algunos autores observan que la inhibición de ERK1/2 en G2 reduce el número de células que entran en mitosis30 o, si es en metafase/anafase o en telofase impide la abscisión celular31, otros aseguran que ERK1/2 no es necesaria para la entrada y salida de las células de mitosis, aunque sí es fundamental para el comienzo de G232. Hayne et al30 han propuesto incluso que p38 MAPK podría inhibir ERK al comienzo de mitosis y permitir así la progresión a través de esta fase.
El papel de las vías de ERK y de p38 MAPK en la entrada en S parece más claro. Ya se ha comentado que la inhibición de p38 MAPK aceleró la entrada en S2 de las células 4n que no podían dividirse por falta de colesterol, lo que conducía a la formación de células poliploides. La inhibición de p38 MAPK en las células en fase G1, con contenido genético 2n, también aceleró su entrada en fase S, como se observó tanto en las células tratadas con SKF 104976 suplementadas con LDL como en las liberadas desde prometafase tras el lavado del nocodazol. Estos efectos subrayan el papel de p38 MAPK reteniendo la replicación del ADN, ya conocido8,11,12,33. Por el contrario, la inhibición de ERK con PD 980359 impidió la entrada en fase S. Este resultado coincide con otros publicados anteriormente por otros autores y señala el papel de ERK para la superación del punto de restricción que antecede la transición de G1 a S34. Por tanto, las rutas de p38 MAPK y de ERK1/2 regulan de manera opuesta la fase de síntesis de ADN, inhibiéndola y favoreciéndola, respectivamente.
En conclusión, los resultados demuestran que la inhibición de la biosíntesis de colesterol impide la citocinesis, lo que hace que las células se acumulen inicialmente en G2/M. Simultáneamente se activa p38 MAPK, que impide la replicación del ADN. No obstante, esta activación de p38 MAPK no se sostiene en el tiempo, probablemente por la deficiencia de p53 que afecta a las células HL-60, lo que conduce a que las células progresen a poliploidía por un mecanismo de endorreplicación. En conjunto, estos resultados ilustran el papel del colesterol en la progresión del ciclo celular y la respuesta de las células ante su deficiencia activando vías de estrés.
Este trabajo ha sido realizado gracias, en parte, a una Beca FEA/SEA 2005 de Investigación Básica y al proyecto SAF2005-07308 del Ministerio de Educación y Ciencia. CIBER Fisiopatología de la Obesidad y Nutrición es una iniciativa del ICSIII. LJF ha recibido una beca de la Fundación Carolina.