




El estrés del retículo endoplasmático (RE) se ha relacionado con distintas enfermedades cardiovasculares, como la arteriosclerosis y la hipertrofia e insuficiencia cardíacas. Este estrés del RE altera la señalización de la insulina, contribuyendo al desarrollo de la resistencia a la insulina y la diabetes. Diversos estudios han demostrado que PPARα inhibe el estrés del RE, por lo que el objetivo de este trabajo consistió en investigar si la activación de este receptor nuclear era capaz de prevenir el estrés del RE inducido por ácidos grasos saturados en células cardíacas, así como los mecanismos implicados.
MétodosCardiomiocitos humanos AC16 fueron tratados con palmitato en presencia de diferentes activadores e inhibidores de AMPK y PPARα. Para los estudios in vivo, ratones macho fueron alimentados con una dieta rica en grasa (HFD). Posteriormente, se determinó la presencia de distintos marcadores de estrés del RE en células cardíacas por medio del análisis de la expresión génica y la acumulación proteica.
ResultadosEl palmitato y la dieta HFD indujeron el estrés del RE en células cardíacas, pues incrementaron diversos marcadores de este, como son la expresión génica de ATF3, BiP/GRP78 y CHOP, el splicing de XBP1 y la fosforilación de IRE-1α y eIF2α. El tratamiento con Wy-14,643, un agonista de PPARα, previno el incremento del estrés del RE inducido por palmitato por medio de la activación de la AMPK.
ConclusiónWy-14,643 podría ser útil para prevenir el estrés del RE y las enfermedades cardiovasculares asociadas en pacientes obesos, e incluso durante la cardiomiopatía diabética, por medio de la activación de AMPK.
Endoplasmic reticulum (ER) stress has been linked to several cardiovascular diseases, such as atherosclerosis, heart failure and cardiac hypertrophy. ER stress impairs insulin signalling, thus contributing to the development of insulin resistance and diabetes. Since several studies have reported that PPARα may inhibit ER stress, the main aim of this study consisted in investigating whether activation of this nuclear receptor is able to prevent lipid-induced ER stress in cardiac cells, as well as studying the mechanisms involved.
MethodsA cardiomyocyte cell line of human origin, AC16, was treated with palmitate in the presence or absence of several AMPK and PPARα pharmacological agonists and antagonists. For the in vivo studies, wild-type male mice were fed a standard diet, or a high-fat diet (HFD), for two months. At the end of the experiments, several ER stress markers were assessed in cardiac cells or in the mice hearts, using real-time RT-PCR and Western-blot analyses.
ResultsThe results demonstrate that both palmitate and the HFD induced ER stress in cardiac cells, since they upregulated the expression (ATF3, BiP/GRP78 and CHOP), splicing (sXBP1), and phosphorylation (IRE-1α and eIF2α) of several ER stress markers. Interestingly, treatment with the PPARα agonist Wy-14,643 prevented an increase in the majority of these ER stress markers in human cardiac cells by means of AMPK activation.
ConclusionThese data indicate that PPARα activation by Wy-14,643 might be useful to prevent the harmful effects of ER stress and associated cardiovascular diseases in obese patients, and even during diabetic cardiomyopathy, by enhancing AMPK activity.
La diabetes de tipo 2 y la obesidad, especialmente si no se corrigen, se encuentran entre los principales factores de riesgo para el desarrollo de enfermedades cardiovasculares. De hecho, la clásica dieta Western, debido a su alto contenido en grasa, presenta diferentes efectos sobre la fisiología cardiovascular que parecen estar relacionados directamente con una amplia variedad de efectos adversos a nivel cardíaco, incluyendo inflamación, hipertrofia, fibrosis y disfunción contráctil1. Sin embargo, los mecanismos a través de los cuales el consumo de dietas ricas en grasas participa en la progresión de estos trastornos son aún poco conocidos. En los pacientes con diabetes de tipo 2 y obesidad es frecuente observar niveles elevados de ácidos grasos libres en plasma, los cuales son responsables de múltiples efectos nocivos sobre el corazón, como la activación del retículo endoplasmático (RE) y procesos inflamatorios crónicos de baja intensidad. Por ejemplo, se han detectado niveles elevados de marcadores clave en el estrés del RE tanto en pacientes diabéticos2 como en ratones diabéticos y obesos3,4. Asimismo, se ha indicado que los ácidos grasos saturados provocan resistencia a la insulina por medio de la inducción del estrés del RE en células β-pancreáticas5,6, hepatocitos7 y células musculares8,9, tanto de origen humano como murino.
El RE es el orgánulo responsable del plegamiento, así como de la correcta maduración, de las proteínas sintetizadas de novo en las células eucariotas. Las proteínas plegadas correctamente viajan desde la luz del RE a otros orgánulos celulares, la membrana celular e incluso el espacio extracelular, mientras que las proteínas mal plegadas son dirigidas hacia la vía de degradación proteosomal asociada al RE (ERAD) para su posterior eliminación10. Cualquier perturbación fisiológica o patológica que interfiera con el normal funcionamiento del RE provocará la acumulación de proteínas plegadas incorrectamente, lo que conducirá a la activación por parte del RE de lo que se conoce como la respuesta a proteínas mal plegadas (UPR)11. La activación de esta UPR implica 3 proteínas de señalización principales: el factor de transcripción activador (ATF)6, la enzima dependiente de inositol (IRE)-1α y la cinasa del RE de tipo PKR (PERK). En ausencia de estrés, los extremos N-terminales transmembrana de estas proteínas se encuentran unidas a la proteína intraluminal BiP/GRP78 (proteína de unión a inmunoglobulina/proteína 78 regulada por glucosa). Ante la presencia de un factor de estrés, el gran exceso de proteínas mal plegadas secuestra BiP/GRP78 de las proteínas transmembrana del RE, lo que inducirá la UPR. En concreto, ATF6 es transportado desde el RE hasta el complejo de Golgi, en el cual una escisión proteolítica libera un fragmento soluble que se trasloca hacia el núcleo, donde actuará como un factor de transcripción para chaperonas del RE y genes relacionados con la vía ERAD12. Por otro lado, la actividad endoribonucleasa de IRE-1α escinde un segmento de 26 pares de bases del ARNm de la proteína de unión a X-box 1 (XBP1), generando un mensaje alternativo que se traduce en una forma activa de este factor de transcripción, XBP1s. Finalmente, PERK fosforila e inhibe el factor de iniciación eucariota 2α (eIF2α), impidiendo de esta manera la traducción de la mayoría de los ARNm10. No obstante, algunos ARNm logran escapar de este control de la traducción, como es el caso de ATF4, un regulador clave de la respuesta del RE al estrés, ya que es capaz de inducir la expresión de ATF3, BiP/GRP78, proteína homóloga a C/EBP (CHOP) o GADD153 y genes implicados en la autofagia, respuesta antioxidante y apoptosis13.
Inicialmente, la activación de la UPR tiene como objetivo principal mitigar los efectos adversos del estrés del RE y, por lo tanto, incrementar la supervivencia celular. Esto se consigue mediante la detención de la traducción general de ARNm, facilitando la degradación de proteínas a través de la vía ERAD y mejorando la producción de chaperonas moleculares implicadas en el plegamiento proteico. Si el estrés del RE es limitado, la UPR potenciará la autofagia para proteger las células14, pero si este estrés no se mitiga dentro de un cierto período, o el trastorno es prolongado, entonces la UPR iniciará el proceso apoptótico con el fin de eliminar las células que amenazan la integridad del organismo15. En el corazón adulto, los cardiomiocitos rara vez proliferan y, como consecuencia, su pérdida debido a la apoptosis puede desempeñar un papel fundamental en la patogénesis de las enfermedades cardiovasculares12. En consonancia con esto, se ha descrito que el estrés del RE está implicado en la patogénesis de la cardiomiopatía diabética como consecuencia del incremento de la muerte celular que provoca en el miocardio de ratas diabéticas16. Igualmente, el miocardio de 2 modelos distintos de rata con diabetes de tipo 2 también muestra estrés del RE17,18. En concreto, en el miocardio de uno de estos modelos, la rata diabética no obesa SDT, se produce un incremento de marcadores de estrés del RE y de señalización pro y antiapoptótica que coincide con una disminución de proteínas relacionadas con el metabolismo de la glucosa y los lípidos17. Entre estas proteínas destacan los receptores activados por proliferadores peroxisómicos (PPAR) de tipo α (PPARα) y γ (PPARγ), además de genes diana de estos, como son los transportadores de glucosa (GLUT4) y ácidos grasos (carnitina palmitoiltransferasa-1 [CPT-1])17. Los PPAR son factores de transcripción dependientes de ligando que pertenecen a la superfamilia de receptores nucleares y que forman heterodímeros con el receptor de retinoides X para unirse al ADN de sus genes diana e iniciar su transcripción génica. De los 3 subtipos de PPAR presentes en mamíferos, PPARα (NR1C1) y PPARγ (NR1C3) son, respectivamente, dianas terapéuticas de fármacos hipolipidemiantes (fibratos) y antidiabéticos (tiazolidinedionas). PPARα se expresa en tejidos con una elevada capacidad de β-oxidación de ácidos grasos, como el hígado y el corazón, donde lleva a cabo funciones de regulación del catabolismo lipídico y de control de la inflamación19-21. Puesto que se ha indicado que la inhibición del estrés del RE podría ser una diana terapéutica útil para la prevención y el tratamiento de la cardiomiopatía diabética, este estudio fue diseñado, en primer lugar, para comprender los mecanismos mediante los cuales la exposición a ácidos grasos saturados resulta en estrés del RE en células cardíacas y, en segundo lugar, investigar si la activación de PPARα podría prevenir este estrés.
Materiales y métodosTécnicas de cultivo celularLas células cardíacas humanas AC16 fueron crecidas y mantenidas tal y como ya se ha descrito previamente22. Brevemente, las células AC16 no diferenciadas fueron mantenidas con medio de cultivo Dulbecco's modified Eagle's medium (DMEM):F12 suplementado con un 12,5% (v:v) de suero bovino fetal, 1% (p:v) de penicilina-estreptomicina y 1% (p:v) de fungizona (Life Technologies, S.A., España), en un incubador a 37°C y 5% de CO2, hasta llegar a un 70-80% de confluencia. Las células fueron entonces estimuladas durante 18h con un medio que contenía palmitato (0,25mM), en presencia o ausencia de distintos agonistas o antagonistas farmacológicos. El medio con palmitato se preparó por conjugación con albúmina sérica bovina (BSA) libre de ácidos grasos23. Después de la incubación y los correspondientes tratamientos, se extrajeron el ARN y las proteínas, tal y como se describe a continuación.
AnimalesRatones C57BL/6X129/SV macho fueron mantenidos bajo condiciones estándar de iluminación (ciclos de 12h de luz/oscuridad) y temperatura (21±1°C), y alimentados con una dieta estándar hasta el comienzo de los estudios, teniendo acceso ad libitum a bebida y alimento durante todo el procedimiento. A los 5 meses de edad, los ratones fueron divididos en 2 grupos experimentales: uno fue alimentado con una dieta estándar (con un 4% de grasa) y el otro con una dieta rica en grasas (high-fat diet [HFD], con un 45% Kcal procedente de grasas, un 91,5% de las cuáles ácidos grasos saturados) durante 8 semanas. La investigación fue realizada de acuerdo con la Guía para el cuidado y el uso de animales de laboratorio publicada por el US National Institutes of Health (Publication n.° 85-23, revisada en 1996). Todos los procedimientos fueron aprobados por el Comité Ético de la Universitat de Barcelona, conforme a lo establecido en la Ley 5/21 de julio de 1995, aprobada por la Generalitat de Catalunya. Al final del tratamiento, los ratones fueron anestesiados con isofluorano 5% y, después de monitorizar la eficacia de la anestesia por medio de la determinación de los reflejos en la pata trasera, fueron sacrificados por dislocación cervical. Después de esto, se extirpó el corazón, que se lavó en tampón fosfato salino frío y finalmente fue congelado en nitrógeno líquido para ser conservado a –80°C hasta su análisis posterior.
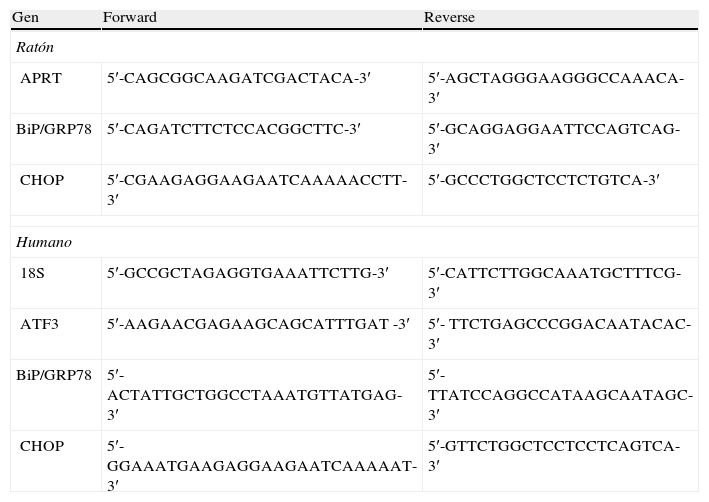
Obtención del ARN y análisis de la expresión génica por reacción en cadena de la polimerasa cuantitativa en tiempo realEl ARN total fue aislado utilizando el reactivo Ultraspec (Biotecx, Houston, EE. UU.), siguiendo las indicaciones del fabricante. Los niveles relativos de ARNm fueron analizados mediante la técnica de transcriptasa reversa asociada a la reacción en cadena de la polimerasa (RT-PCR) a tiempo real, tal y como se ha descrito previamente24. Los niveles de XBP1s se determinaron por electroforesis en gel de agarosa8. Las secuencias de los oligonucleótidos utilizados para la amplificación se describen en la tabla 1.
Oligonucleótidos utilizados para los análisis de RT-PCR cuantitativa en tiempo real
| Gen | Forward | Reverse |
| Ratón | ||
| APRT | 5′-CAGCGGCAAGATCGACTACA-3′ | 5′-AGCTAGGGAAGGGCCAAACA-3′ |
| BiP/GRP78 | 5′-CAGATCTTCTCCACGGCTTC-3′ | 5′-GCAGGAGGAATTCCAGTCAG-3′ |
| CHOP | 5′-CGAAGAGGAAGAATCAAAAACCTT-3′ | 5′-GCCCTGGCTCCTCTGTCA-3′ |
| Humano | ||
| 18S | 5′-GCCGCTAGAGGTGAAATTCTTG-3′ | 5′-CATTCTTGGCAAATGCTTTCG-3′ |
| ATF3 | 5′-AAGAACGAGAAGCAGCATTTGAT -3′ | 5′- TTCTGAGCCCGGACAATACAC-3′ |
| BiP/GRP78 | 5′-ACTATTGCTGGCCTAAATGTTATGAG-3′ | 5′-TTATCCAGGCCATAAGCAATAGC-3′ |
| CHOP | 5′-GGAAATGAAGAGGAAGAATCAAAAAT-3′ | 5′-GTTCTGGCTCCTCCTCAGTCA-3′ |
Los extractos proteicos totales fueron obtenidos por lisis de las células en tampón RIPA (Sigma, St Louis, EE. UU.) con inhibidores de fosfatasas y proteasas. El homogenizado resultante fue centrifugado a 10.000g durante 30 min a 4°C, y la concentración proteica contenida en el sobrenadante fue determinada mediante el método de Bradford25, adaptado a microplacas. Posteriormente, las proteínas fueron separadas en geles de SDS-PAGE al 10% (p:v) de acrilamida y transferidas a membranas de polivilideno (Millipore, Bedford, EE. UU.). Para la determinación de IRE-1α se utilizó un gel phos-tag según se ha descrito en la bibliografía26. La inmunodetección se realizó utilizando anticuerpos específicos (Cell Signaling Technology, Danvers, EE. UU.; Sigma) y el kit de quimioluminiscencia Western Lightning® Plus-ECL (PerkinElmer, Waltham, EE. UU.). El tamaño de las proteínas se determinó utilizando un estándar de pesos moleculares (Life Technologies).
Análisis estadísticosLos resultados se expresan como la media±desviación estándar de al menos 3 experimentos independientes para los estudios in vitro, cada uno de ellos consistente en 3 placas de cultivo (n=9), y 5 ratones para los experimentos in vivo. Las diferencias significativas fueron establecidas mediante los tests estadísticos t de Student o ANOVA, dependiendo del número de grupos comparados, y utilizando el programa informático GaphPad Prism (GraphPad Software Inc, San Diego, EE. UU.). En los casos en que se encontraron diferencias estadísticamente significativas, se aplicó el post-test de Tukey-Kramer para comparaciones múltiples. Las diferencias fueron consideradas significativas a partir de p<0,05.
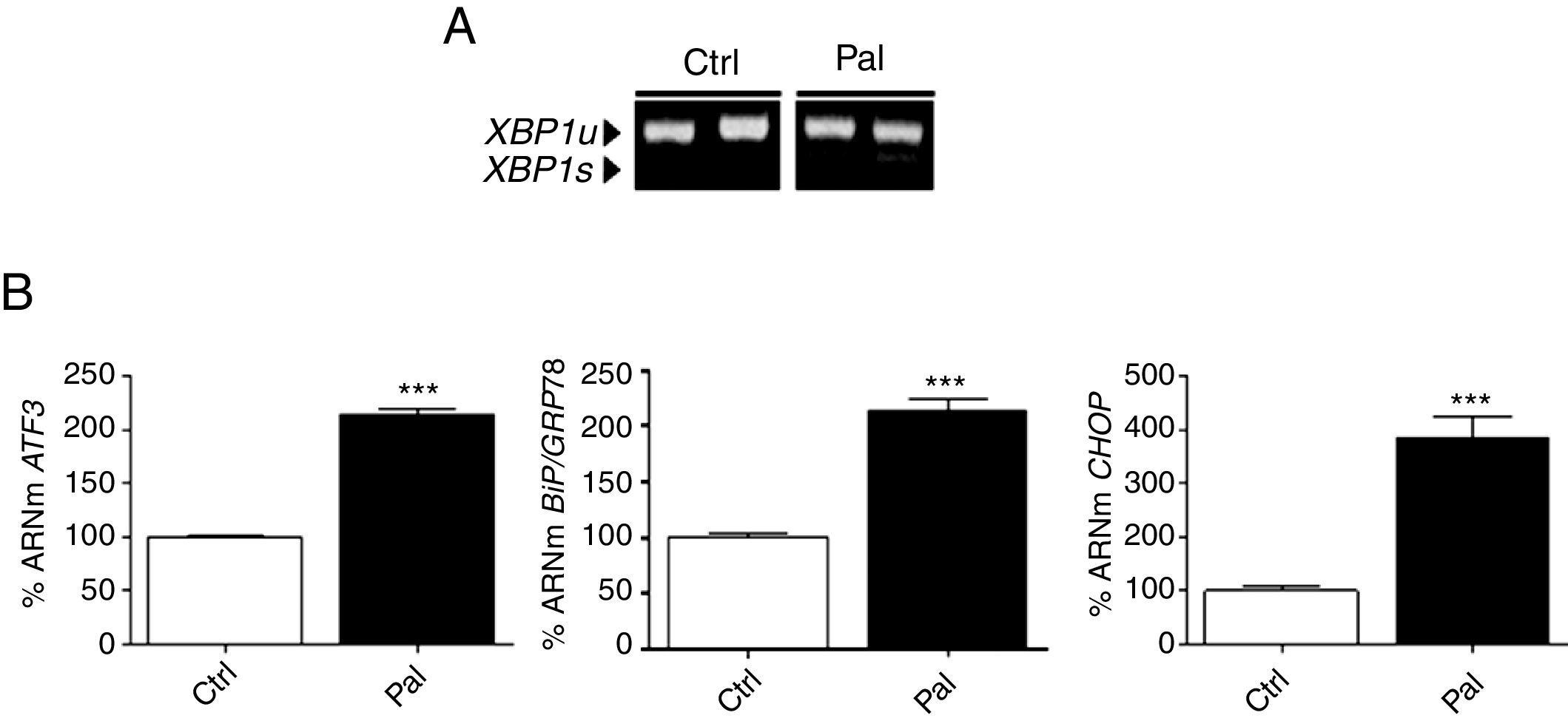
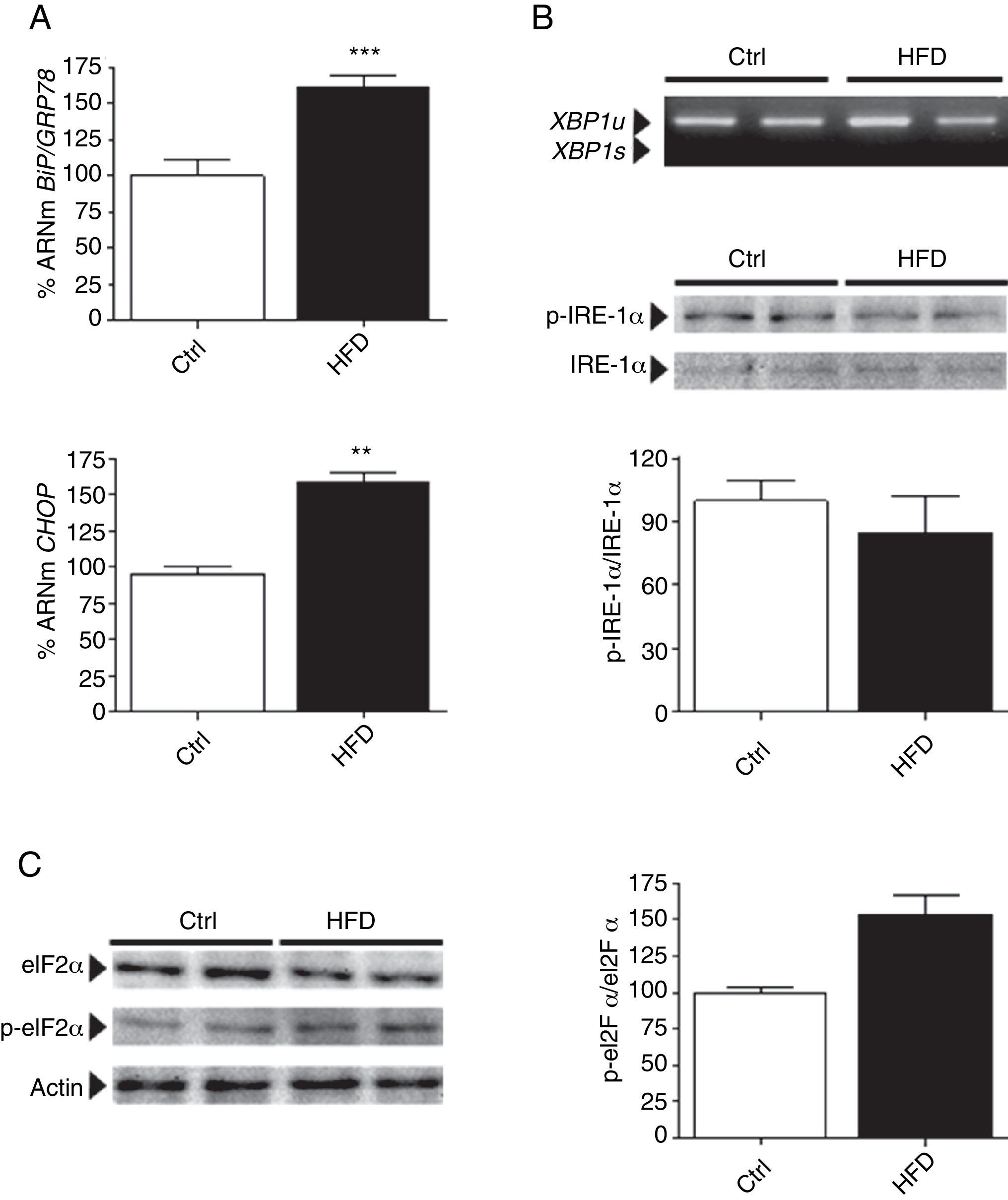
ResultadosLos ácidos grasos saturados incrementan los marcadores de estrés del retículo endoplasmático en células cardíacasEn primer lugar, se determinó si el ácido graso saturado palmitato era capaz de inducir la expresión de distintos marcadores de estrés del RE en células cardíacas humanas AC16. Como se observa en la figura 1 A, el palmitato indujo el splicing, y por lo tanto la activación, de XBP1, en comparación con las células expuestas solo a BSA. Asimismo, los análisis de RT-PCR cuantitativa en tiempo real demostraron que este ácido graso saturado inducía de manera significativa la expresión de ATF3 (aproximadamente 2 veces, p<0,001 vs. control), BiP/GRP78 (∼ 2 veces, p<0,001) y CHOP (∼ 4 veces, p<0,001) (fig. 1 B). Para corroborar los resultados obtenidos in vitro con células cardíacas humanas, también se realizaron estudios in vivo con ratones alimentados con dietas ricas en ácidos grasos saturados (HFD). De acuerdo con lo observado in vitro, la dieta HFD indujo de manera significativa la expresión de BiP/GRP78 y CHOP en el corazón de estos ratones (∼ 1,5 veces, p<0,001 y p<0,01, respectivamente, vs. dieta control) (fig. 2 A). Por el contrario, la administración de una dieta rica en grasas no incrementó el splicing de XBP1 ni, en consonancia con esto, tampoco modificó la fosforilación de IRE-1α (fig. 2 B), pero sí indujo la fosforilación de eIF2α en Ser51 (fig. 2 C).
 Niveles de XBP1u/XBP1s (unspliced/spliced) determinados mediante RT-PCR y B) cuantificación relativa de los niveles de ARNm de ATF3, CHOP y BiP/GRP78 determinados mediante RT-PCR cuantitativa en tiempo real, de muestras obtenidas de células AC16 tratadas con palmitato (Pal, 0,25mM, 18h). Los gráficos representan los niveles de ARNm normalizados con el gen control 18S y están expresados como porcentaje respecto el grupo control (Ctrl)±DE. ***p<0,001 vs. Ctrl.")
El palmitato induce el estrés del RE en cardiomiocitos AC16. A) Niveles de XBP1u/XBP1s (unspliced/spliced) determinados mediante RT-PCR y B) cuantificación relativa de los niveles de ARNm de ATF3, CHOP y BiP/GRP78 determinados mediante RT-PCR cuantitativa en tiempo real, de muestras obtenidas de células AC16 tratadas con palmitato (Pal, 0,25mM, 18h). Los gráficos representan los niveles de ARNm normalizados con el gen control 18S y están expresados como porcentaje respecto el grupo control (Ctrl)±DE.
***p<0,001 vs. Ctrl.
![La dieta rica en grasas (HFD) induce el estrés del RE en corazón de ratón. Cuantificación relativa de los niveles de: A) ARNm de BiP/GRP78 y CHOP, determinados mediante RT-PCR cuantitativa en tiempo real, y B) XBP1u/XBP1s (unspliced/spliced), determinados mediante RT-PCR, de muestras obtenidas de corazón de ratones alimentados con un dieta estándar (control [Ctrl]) o rica en ácidos grasos saturados (HFD) durante 8 semanas. Los gráficos representan los niveles de ARNm normalizados con el gen control APRT y están expresados como porcentaje respecto el grupo control±DE. C) Análisis por Western-blot de los niveles de proteína IRE-1α total y fosforilada, y eIF2α total y fosforilada en las mismas muestras descritas en el panel A. Los gráficos representan la cuantificación de los niveles de proteína normalizados y están expresados como porcentaje de las muestras control±DE. Todas las autorradiografías son representativas de 2 experimentos independientes. *p<0,05 vs. Ctrl. ** p<0,01 vs. Ctrl. ***p<0,001 vs. Ctrl.](https://static.elsevier.es/multimedia/02149168/0000002600000006/v1_201411160049/S0214916814000394/v1_201411160049/es/main.assets/gr2.jpeg?xkr=ue/ImdikoIMrsJoerZ+w997EogCnBdOOD93cPFbanNcxT+B4FjbiPdoCJto3d5S/Ch6vvG+Cp80/Jya7nfY0UvLkSpgsF6pdUV/dKgWQV8qLEQhdOYA3HJgNoBV9y5Ppc15hsfBPW+NP4rRr7D2AISqARFMVWmEvdo2pTx4kZp9ax26HebEtw0duEn+bjwwihbzKwmMKJbnTxwMNpICkgahIZV+o4negYOXNHahyMn/uG50JxeDuad2rXn+vpEX+duLUQqe6bOqOplCCZENxccUC2K0jZlbq13U6h2rL3Qm3OYdshCzGb52KHUIzeZJa "La dieta rica en grasas (HFD) induce el estrés del RE en corazón de ratón. Cuantificación relativa de los niveles de: A) ARNm de BiP/GRP78 y CHOP, determinados mediante RT-PCR cuantitativa en tiempo real, y B) XBP1u/XBP1s (unspliced/spliced), determinados mediante RT-PCR, de muestras obtenidas de corazón de ratones alimentados con un dieta estándar (control [Ctrl]) o rica en ácidos grasos saturados (HFD) durante 8 semanas. Los gráficos representan los niveles de ARNm normalizados con el gen control APRT y están expresados como porcentaje respecto el grupo control±DE. C) Análisis por Western-blot de los niveles de proteína IRE-1α total y fosforilada, y eIF2α total y fosforilada en las mismas muestras descritas en el panel A. Los gráficos representan la cuantificación de los niveles de proteína normalizados y están expresados como porcentaje de las muestras control±DE. Todas las autorradiografías son representativas de 2 experimentos independientes. *p<0,05 vs. Ctrl. ** p<0,01 vs. Ctrl. ***p<0,001 vs. Ctrl.")
La dieta rica en grasas (HFD) induce el estrés del RE en corazón de ratón. Cuantificación relativa de los niveles de: A) ARNm de BiP/GRP78 y CHOP, determinados mediante RT-PCR cuantitativa en tiempo real, y B) XBP1u/XBP1s (unspliced/spliced), determinados mediante RT-PCR, de muestras obtenidas de corazón de ratones alimentados con un dieta estándar (control [Ctrl]) o rica en ácidos grasos saturados (HFD) durante 8 semanas. Los gráficos representan los niveles de ARNm normalizados con el gen control APRT y están expresados como porcentaje respecto el grupo control±DE. C) Análisis por Western-blot de los niveles de proteína IRE-1α total y fosforilada, y eIF2α total y fosforilada en las mismas muestras descritas en el panel A. Los gráficos representan la cuantificación de los niveles de proteína normalizados y están expresados como porcentaje de las muestras control±DE. Todas las autorradiografías son representativas de 2 experimentos independientes.
*p<0,05 vs. Ctrl.
** p<0,01 vs. Ctrl.
***p<0,001 vs. Ctrl.
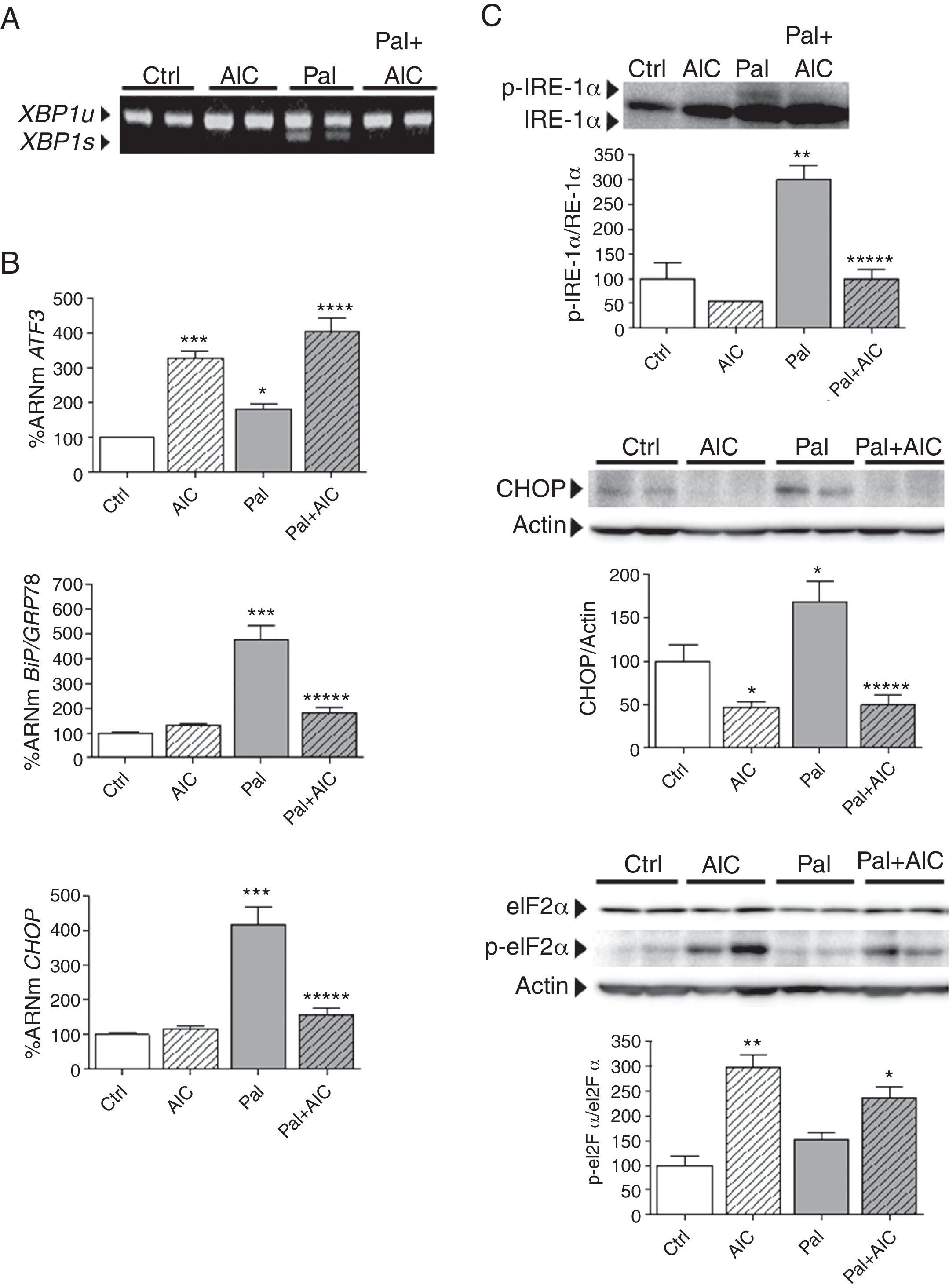
Diversos estudios han demostrado que la inducción de la proteína cinasa activada por 5’-AMP (AMPK), la cual es considerada una diana farmacológica para el tratamiento de la resistencia a la insulina y la diabetes de tipo 227, inhibe el estrés del RE8,28, mientras que la reducción de la actividad de esta cinasa lo promueve29. En consecuencia, nuestro siguiente objetivo fue investigar el papel de la AMPK en el estrés del RE inducido por palmitato en células cardíacas humanas. Para ello, utilizamos un mimético de AMP que actúa como activador de la AMPK, el 5-aminoimidazol-4-carboxamida ribonucleótido (AICAR), así como un inhibidor de esta, el Compuesto C. Tal y como demuestran nuestros resultados, AICAR redujo el splicing de XBP1 inducido por palmitato (fig. 3 A), así como la expresión de BiP/GRP78 y CHOP, aunque incrementó la expresión de ATF3 (∼ 3,5 veces, p<0,001 vs. control, fig. 3 B). AICAR también previno el incremento inducido por palmitato de la proteína CHOP y la activación por fosforilación de IRE-1α (fig. 3 C), cuya actividad endoribonucleasa provoca el splicing de XBP1. Estos resultados concuerdan con la reducción de XBP1s y de la transcripción de CHOP.
 en presencia o ausencia de AICAR (AIC, 2mM, 24h). A) Niveles de XBP1u/XBP1s (unspliced/spliced) determinados mediante RT-PCR. B) Cuantificación relativa de los niveles de ARNm de ATF3, CHOP y BiP/GRP78 determinados mediante RT-PCR cuantitativa en tiempo real. Los gráficos representan los niveles de ARNm normalizados con el gen control 18S y están expresados como porcentaje respecto del grupo control (Ctrl)±DE. C) Análisis por Western-blot de los niveles de proteína IRE-1α total y fosforilada, CHOP, y eIF2α total y fosforilada en Ser51. Los gráficos representan la cuantificación de los niveles de proteína normalizados y están expresados como porcentaje de las muestras control±DE. Todas las autorradiografías son representativas de 2 experimentos independientes. *p<0,05 vs. Ctrl. **p<0,01 vs. Ctrl. ***p<0,001 vs. Ctrl. ****p<0,05 vs. Pal. *****p<0,01 vs. Pal.")
La activación de AMPK previene el estrés del RE inducido por palmitato en células cardíacas humanas. Células AC16 fueron tratadas con palmitato (Pal, 0,25mM, 18h) en presencia o ausencia de AICAR (AIC, 2mM, 24h). A) Niveles de XBP1u/XBP1s (unspliced/spliced) determinados mediante RT-PCR. B) Cuantificación relativa de los niveles de ARNm de ATF3, CHOP y BiP/GRP78 determinados mediante RT-PCR cuantitativa en tiempo real. Los gráficos representan los niveles de ARNm normalizados con el gen control 18S y están expresados como porcentaje respecto del grupo control (Ctrl)±DE. C) Análisis por Western-blot de los niveles de proteína IRE-1α total y fosforilada, CHOP, y eIF2α total y fosforilada en Ser51. Los gráficos representan la cuantificación de los niveles de proteína normalizados y están expresados como porcentaje de las muestras control±DE. Todas las autorradiografías son representativas de 2 experimentos independientes.
*p<0,05 vs. Ctrl.
**p<0,01 vs. Ctrl.
***p<0,001 vs. Ctrl.
****p<0,05 vs. Pal.
*****p<0,01 vs. Pal.
Por el contrario, AICAR indujo la fosforilación de eIF2α en Ser51 (∼ 3 veces, p<0,01 vs. control, fig. 3 C). El significado fisiológico del incremento de la fosforilación de eIF2α en Ser51 permanece aún por elucidar, pues es ampliamente conocido que la fosforilación de eIF2α inhibe la traducción genérica del ARNm a proteína en el RE, pero podría estar relacionado con los efectos descritos para AICAR sobre la expresión de ATF3 ya descritos en la figura 3 B. Este papel dual de AICAR ya había sido descrito previamente en cardiomiocitos de ratón HL-1, donde este activador de AMPK reduce la apoptosis inducida por hipoxia/reoxigenación10. En todos nuestros estudios, la activación de AMPK por AICAR se confirmó mediante Western-blot por determinación de la fosforilación de AMPK en Thr172, la cual es esencial para su actividad, así como la fosforilación de una proteína diana de esta, la acetil-CoA carboxilasa 2 (ACC2) en Ser218 (datos no mostrados).
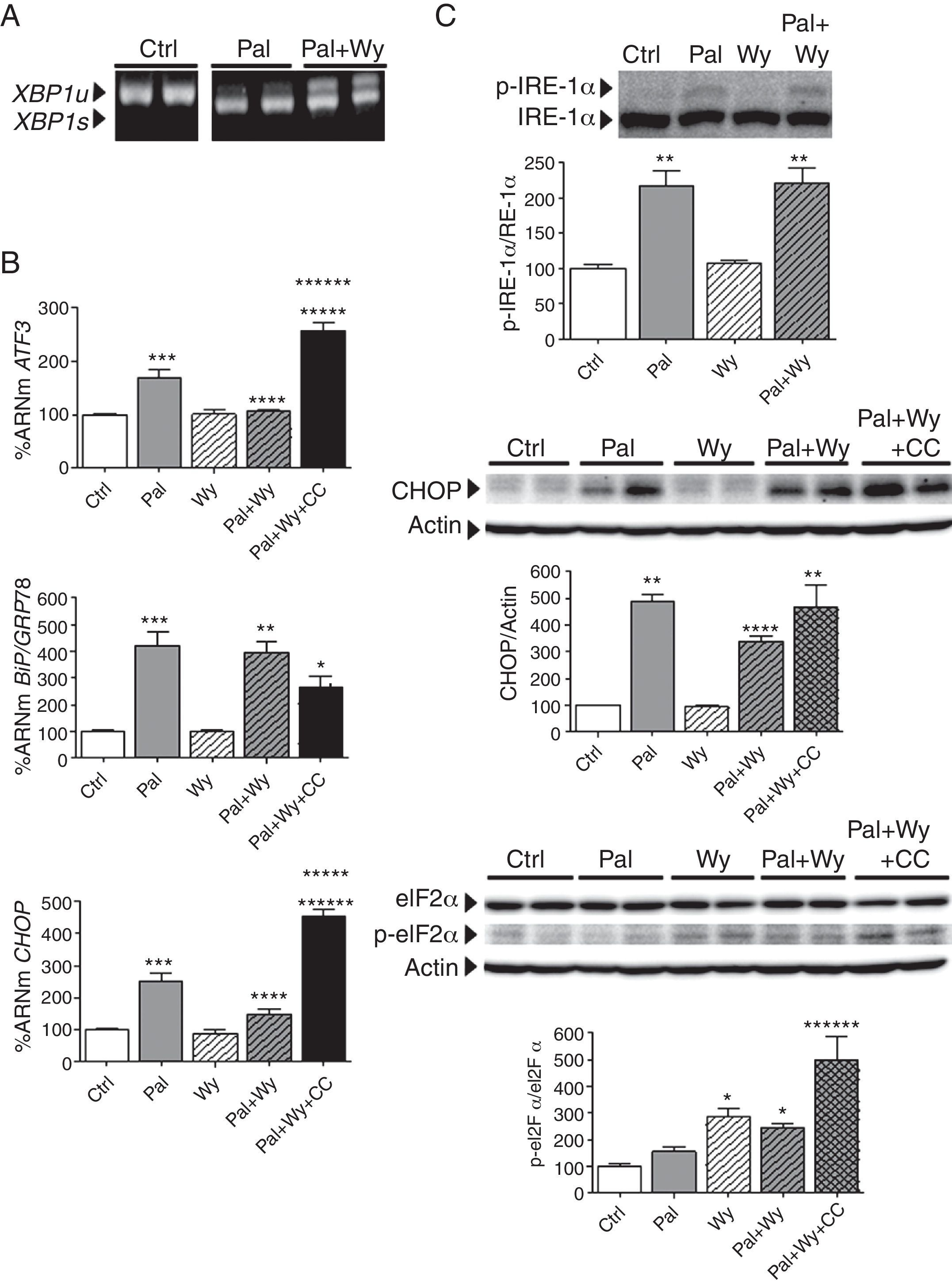
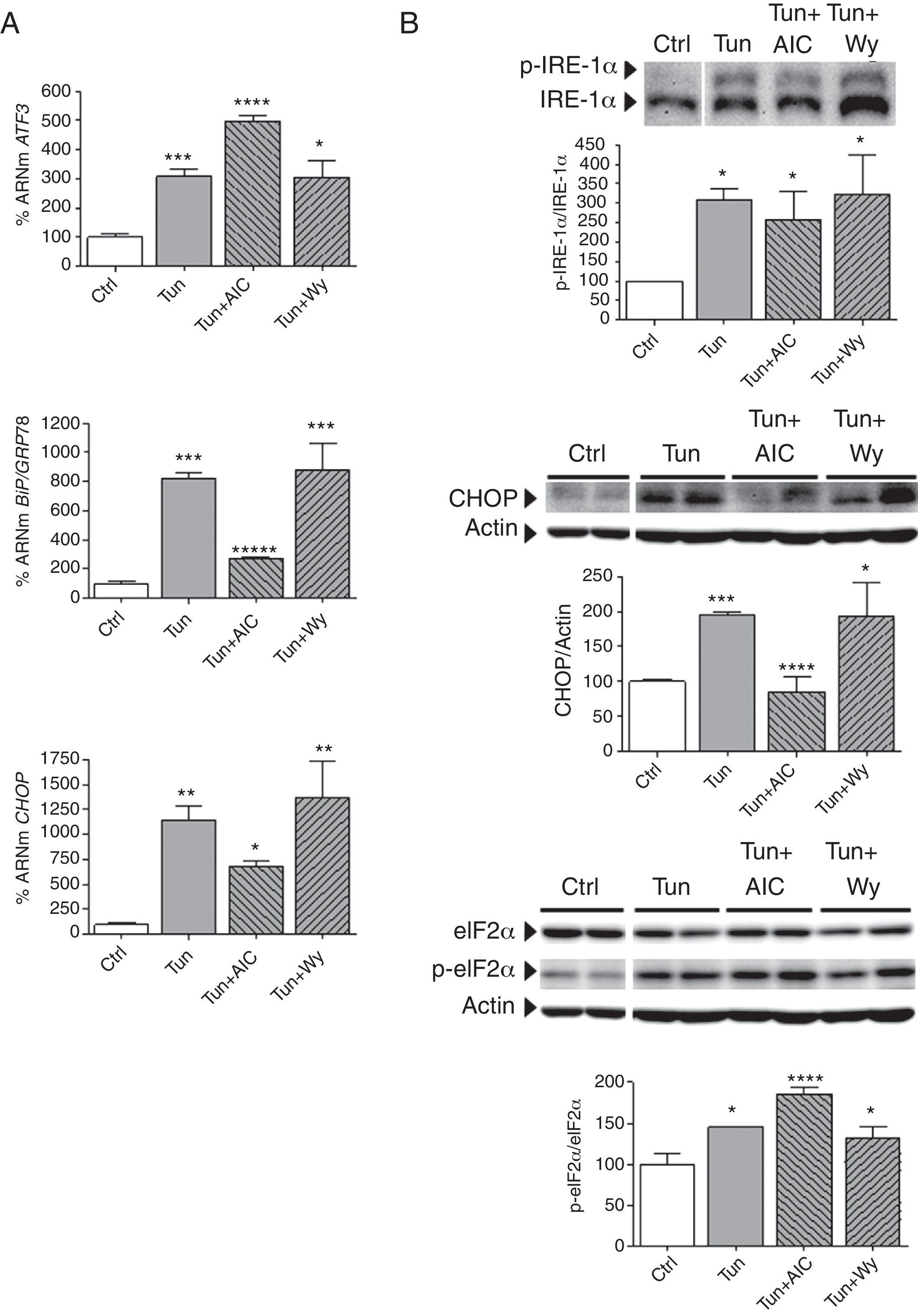
La activación de la proteína cinasa activada por 5’-AMP inducida por Wy-14,643 atenúa el estrés del retículo endoplasmático inducido por palmitato pero no por tunicamicinaPPARα es un factor de transcripción clave para el control del metabolismo celular. Dado que la AMPK es también un regulador importante de la homeóstasis metabólica que puede ser inducida por agonistas de PPARα, y que la desregulación de PPARα tiene un papel importante en el desarrollo de la hipertrofia y la insuficiencia cardíacas, a continuación investigamos el papel de un agonista de PPARα, Wy-14,643 (10μM, 24h), sobre el estrés del RE inducido por palmitato en células cardíacas humanas. Como se observa en la figura 4, el tratamiento con Wy-14,643 redujo los niveles de expresión de ATF3 y CHOP inducidos por palmitato, pero no previno el splicing de XBP1, ni tampoco el importante incremento de expresión de BiP/GRP78. La coincubación con un inhibidor de AMPK, el Compuesto C, indicaba que los efectos beneficiosos de Wy-14,643 sobre ATF3 y CHOP eran mediados por la activación de AMPK. Al mismo tiempo, el tratamiento con el Compuesto C solo no modificaba ninguno de estos genes (datos no mostrados). Estos cambios en la expresión génica de marcadores de estrés del RE coincidieron con los niveles proteicos determinados por Western-blot. Así, la figura 4 C muestra cómo el cotratamiento con Wy-14,643 no revirtió la fosforilación de IRE-1α inducida por palmitato, de la misma manera que tampoco prevenía el splicing de XBP1, demostrando que este agonista de PPARα era incapaz de inhibir la actividad endoribonucleasa de IRE-1α sobre XBP1. Asimismo, y de acuerdo con los niveles de expresión génica, el tratamiento con Wy-14,643 atenuó el incremento en los niveles de proteína CHOP provocado por el palmitato. Esta reducción de los niveles de proteína CHOP inducidos por Wy-14,643 fueron completamente prevenidos en presencia del Compuesto C, demostrando la implicación, una vez más, de la AMPK en los efectos beneficiosos de la activación de PPARα sobre el estrés del RE inducido por palmitato. Sorprendentemente, Wy-14,643 también indujo la fosforilación de eIF2α en Ser51 de manera significativa (∼ 3 veces, p<0,05 vs. control). A diferencia de lo que ocurría con el activador de AMPK, AICAR, este incremento en la fosforilación de eIF2α inducido por activación de PPARα no correlacionaba con una mayor expresión de ATF3.
 en presencia o ausencia de Wy-14,643 (Wy, 10μM, 24h) y del Compuesto C (CC, 30μM, 25h). A) Niveles de XBP1u/XBP1s (unspliced/spliced) determinados mediante RT-PCR. B) Cuantificación relativa de los niveles de ARNm de ATF3, CHOP y BiP/GRP78 determinados mediante RT-PCR cuantitativa en tiempo real. Los gráficos representan los niveles de ARNm normalizados con el gen control 18S y están expresados como porcentaje respecto del grupo control (Ctrl)±DE. C) Análisis por Western-blot de los niveles de proteína IRE-1α total y fosforilada, CHOP, y eIF2α total y fosforilada en Ser51. Los gráficos representan la cuantificación de los niveles de proteína normalizados y están expresados como porcentaje de las muestras control±DE. Todas las autorradiografías son representativas de 2 experimentos independientes. *p<0,05 vs. Ctrl. **p<0,01 vs. Ctrl. ***p<0,001 vs. Ctrl. ****p<0,05 vs. Pal. *****p<0,01 vs. Pal. ******p<0,01 vs. Pal+Wy.")
Efecto de la activación de PPARα con Wy-14,643 sobre el estrés del RE. Células AC16 fueron tratadas con palmitato (Pal, 0,25mM, 18h) en presencia o ausencia de Wy-14,643 (Wy, 10μM, 24h) y del Compuesto C (CC, 30μM, 25h). A) Niveles de XBP1u/XBP1s (unspliced/spliced) determinados mediante RT-PCR. B) Cuantificación relativa de los niveles de ARNm de ATF3, CHOP y BiP/GRP78 determinados mediante RT-PCR cuantitativa en tiempo real. Los gráficos representan los niveles de ARNm normalizados con el gen control 18S y están expresados como porcentaje respecto del grupo control (Ctrl)±DE. C) Análisis por Western-blot de los niveles de proteína IRE-1α total y fosforilada, CHOP, y eIF2α total y fosforilada en Ser51. Los gráficos representan la cuantificación de los niveles de proteína normalizados y están expresados como porcentaje de las muestras control±DE. Todas las autorradiografías son representativas de 2 experimentos independientes.
*p<0,05 vs. Ctrl.
**p<0,01 vs. Ctrl.
***p<0,001 vs. Ctrl.
****p<0,05 vs. Pal.
*****p<0,01 vs. Pal.
******p<0,01 vs. Pal+Wy.
La inhibición del estrés del RE después de la activación de la AMPK con AICAR también se demostró en células cardíacas inducidas con tunicamicina, una mezcla de antibióticos nucleósidos homólogos que actúa como un potente inductor farmacológico del estrés del RE. Como era de esperar, la tunicamicina provocó un aumento muy importante en la expresión de distintos marcadores de estrés, como son el ATF3 (∼ 3 veces, p<0,001 vs. control), BiP/GRP78 (∼ 8 veces, p<0,001) y CHOP (∼ 11 veces, p<0,001), además de inducir los niveles proteicos de CHOP (∼ 2 veces, p<0,001) y la fosforilación de IRE-1α (∼ 3 veces, p<0,05) y eIF2α (∼ 1,5 veces, p<0,05) (fig. 5). La adición de AICAR a las células tratadas con tunicamicina impidió el aumento de algunos de estos marcadores, tales como la expresión génica de BiP/GRP78 y CHOP, pero no de ATF3 (fig. 5 A). Los análisis de Western-blot revelaron que AICAR también suprimía el aumento de la proteína CHOP inducido por tunicamicina, pero no de la fosforilación de eIF2α o IRE-1α (fig. 5 B). La activación de AMPK por AICAR se confirmó de nuevo mediante la determinación de la fosforilación de AMPK y ACC2 (datos no mostrados). Por el contrario, la adición de Wy-14,643 a las células tratadas con tunicamicina fue incapaz de prevenir ninguno de los cambios inducidos por tunicamicina sobre estos marcadores (fig. 5).
 en presencia o ausencia de AICAR (AIC, 2mM, 24h) o Wy-14,643 (Wy, 10μM, 24h). A) Cuantificación relativa de los niveles de ARNm de ATF3, CHOP y BiP/GRP78 determinados mediante RT-PCR cuantitativa en tiempo real. Los gráficos representan los niveles de ARNm normalizados con el gen control 18S y están expresados como porcentaje respecto del grupo control (Ctrl)±DE. B) Análisis por Western-blot de los niveles de proteína IRE-1α total y fosforilada, CHOP, y eIF2α total y fosforilada en Ser51. Los gráficos representan la cuantificación de los niveles de proteína normalizados y están expresados como porcentaje de las muestras control±DE. Todas las autorradiografías son representativas de 2 experimentos independientes. *p<0,05 vs. Ctrl. **p<0,01 vs. Ctrl. ***p<0,001 vs. Ctrl. ****p<0,05 vs. Tun. *****p<0,01 vs. Tun.")
AICAR, pero no Wy-14,643, previene el estrés del RE inducido por tunicamicina. Células AC16 fueron tratadas durante 4h con tunicamicina (Tun, 5μg/ml) en presencia o ausencia de AICAR (AIC, 2mM, 24h) o Wy-14,643 (Wy, 10μM, 24h). A) Cuantificación relativa de los niveles de ARNm de ATF3, CHOP y BiP/GRP78 determinados mediante RT-PCR cuantitativa en tiempo real. Los gráficos representan los niveles de ARNm normalizados con el gen control 18S y están expresados como porcentaje respecto del grupo control (Ctrl)±DE. B) Análisis por Western-blot de los niveles de proteína IRE-1α total y fosforilada, CHOP, y eIF2α total y fosforilada en Ser51. Los gráficos representan la cuantificación de los niveles de proteína normalizados y están expresados como porcentaje de las muestras control±DE. Todas las autorradiografías son representativas de 2 experimentos independientes.
*p<0,05 vs. Ctrl.
**p<0,01 vs. Ctrl.
***p<0,001 vs. Ctrl.
****p<0,05 vs. Tun.
*****p<0,01 vs. Tun.
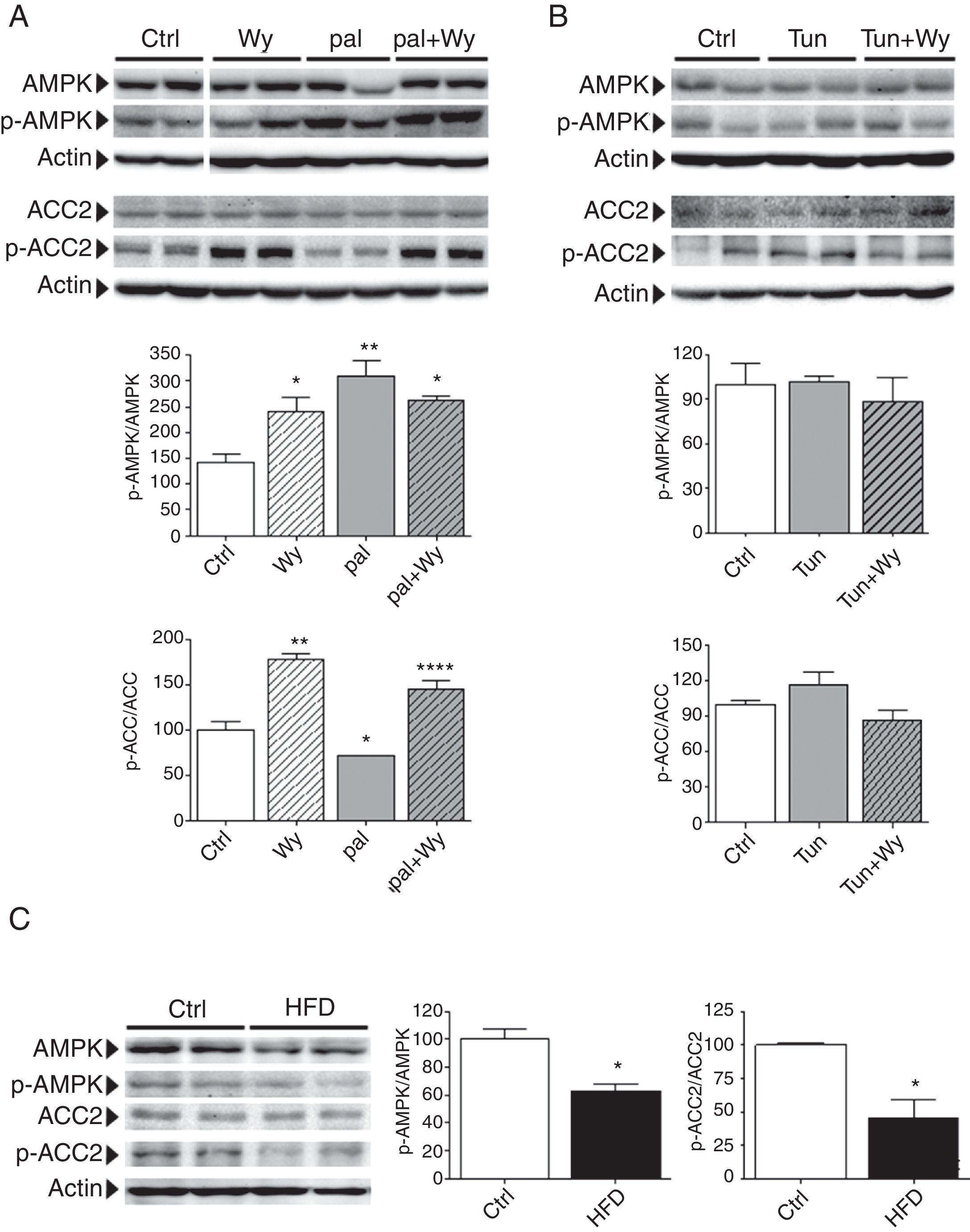
Dado que los agonistas de PPARα, incluyendo Wy-14,643, pueden inducir la fosforilación de AMPK de manera independiente de la activación de PPARα, nuestro último objetivo fue examinar si esta cinasa estaba realmente implicada en los efectos de Wy-14,643. Para ello, monitorizamos los niveles de fosforilación de AMPK en Thr172 mediante Western-blot con anticuerpos específicos. Sorprendentemente, tanto el palmitato (∼ 3 veces, p<0,05 vs. grupo control), como Wy-14,643 (∼ 2,5 veces, p<0,05), indujeron la fosforilación de AMPK en células cardíacas humanas (fig. 6 A). Dado que la regulación de la actividad de AMPK es compleja, y su estado de fosforilación es controlado tanto por fosfatasas como por cinasas, también se exploró la fosforilación de la isoforma ACC2 en Ser218. En contraste con lo que se observaba en la fosforilación de la AMPK, los niveles de fosfo-ACC2 se redujeron ligera pero significativamente en células expuestas a palmitato, mientras que en presencia de Wy-14,643 se incrementaban de manera significativa (∼ 2 veces, p<0,01 vs. control). Por el contrario, Wy-14,643 no incrementó la actividad de AMPK ni modificó la fosforilación de ACC2 en las células tratadas con tunicamicina, proporcionando quizá una explicación plausible al hecho de que Wy-14,643 fuera incapaz de prevenir el estrés del RE inducido por tunicamicina, y reforzando aún más el papel de la AMPK en los efectos positivos de este agonista frente el palmitato. Finalmente, los resultados obtenidos en cardiomiocitos in vitro con el palmitato concuerdan con los datos obtenidos in vivo, donde se detectó una disminución tanto de la fosforilación de AMPK (reducción del 40% vs. ratones control, p<0,01), como de la ACC2 (50% vs. ratones control, p<0,05), en el corazón de los ratones alimentados con una dieta HFD.
 células AC16 tratadas con palmitato (Pal, 0,25mM, 18h) en presencia o ausencia de Wy-14,643 (Wy, 10μM, 24h); B) células AC16 tratadas durante 4h con tunicamicina (Tun, 5μg/ml) en presencia o ausencia de AICAR (AIC, 2mM, 24h) o Wy-14,643 (Wy, 10μM, 24h), y C) corazón de ratones alimentados con un dieta estándar (Ctrl) o rica en ácidos grasos saturados (HFD) durante 8 semanas. Los gráficos representan la cuantificación de los niveles de proteína normalizados en relación con la actina y están expresados como porcentaje de las muestras control (Ctrl)±DE. Todas las autorradiografías son representativas de 2 experimentos independientes. *p < 0,05 vs. Ctrl. **p < 0,01 vs. Ctrl. ***p < 0,001 vs. Ctrl. ****p < 0,05 vs.Pal.")
La activación de AMPK interviene en los efectos de Wy-14,643 sobre el estrés del RE inducido por palmitato. Análisis por Western-blot de los niveles de proteína AMPK total y fosforilada en Thr172, y ACC2 total y fosforilada en Ser218 en muestras obtenidas de: A) células AC16 tratadas con palmitato (Pal, 0,25mM, 18h) en presencia o ausencia de Wy-14,643 (Wy, 10μM, 24h); B) células AC16 tratadas durante 4h con tunicamicina (Tun, 5μg/ml) en presencia o ausencia de AICAR (AIC, 2mM, 24h) o Wy-14,643 (Wy, 10μM, 24h), y C) corazón de ratones alimentados con un dieta estándar (Ctrl) o rica en ácidos grasos saturados (HFD) durante 8 semanas. Los gráficos representan la cuantificación de los niveles de proteína normalizados en relación con la actina y están expresados como porcentaje de las muestras control (Ctrl)±DE. Todas las autorradiografías son representativas de 2 experimentos independientes.
*p < 0,05 vs. Ctrl.
**p < 0,01 vs. Ctrl.
***p < 0,001 vs. Ctrl.
****p < 0,05 vs.Pal.
En los últimos años, la activación de la UPR durante el estrés del RE ha captado la atención de numerosos investigadores como un nuevo mecanismo implicado en la asociación entre la inflamación provocada por los ácidos grasos saturados y las enfermedades metabólicas crónicas, como por ejemplo la obesidad, la resistencia a la insulina y la diabetes de tipo 211,30. Diversos estudios realizados en músculo8,31 y células β pancreáticas32 han demostrado que el palmitato induce el splicing de XBP1 e incrementa la expresión de distintos marcadores de estrés del RE, como ATF3, BiP/GRP78 y CHOP, además de la fosforilación de IRE-1α8,31. De acuerdo con esto, nuestro estudio muestra que el palmitato induce el splicing de XBP1 y la fosforilación de IRE-1α, así como el incremento en la expresión o la acumulación proteica de ATF3, BiP/GRP78 y CHOP, en células cardíacas humanas. El incremento en los niveles de BiP/GRP78 indica que el estrés del RE inducido por palmitato en células cardíacas se corresponde con la fase adaptativa de la UPR, pues esta se caracteriza por un incremento de la expresión de proteínas con función chaperona, como es el caso de BiP/GRP78, que colaboran en la estabilización de los intermediarios de plegamiento proteico33. No obstante, y al contrario de lo descrito previamente en células musculares8,9,34, la vía PERK/eIF2α de la UPR no se activaba en presencia de palmitato, ya que no se observaron cambios en la fosforilación de eIF2α en Ser51. Además, en este estudio demostramos por primera vez en células cardíacas humanas cómo la activación de PPARα con Wy-14,643 previene el estrés del RE inducido por ácidos grasos saturados, pues este agonista evitaba el incremento de ATF3 y CHOP. De manera análoga a lo que sucede en células β pancreáticas tratadas con agonistas de PPARβ/δ35, los efectos beneficiosos de Wy-14,643 sobre nuestras células no derivaba de cambios en los niveles proteicos de la chaperona BiP/GRP78. Asimismo, Wy-14,643 fue incapaz de prevenir la fosforilación de IRE-1α y el splicing de XBP1. No obstante, en relación con este último resultado, no se puede descartar que el tiempo de tratamiento utilizado fuera insuficiente para detectar estos posibles cambios, especialmente teniendo en cuenta que XBP1s es una proteína muy inestable la transcripción de la cual depende tanto de ATF6 como del propio IRE-1α36. XBP1s regula muchos de los procesos esenciales de la UPR implicados en plegamiento proteico, biogénesis de orgánulos, vía ERAD y autofagia, si bien sus dianas génicas pueden variar en función del tipo celular y la naturaleza del agente inductor del estrés10. Aunque la expresión de BiP/GRP78 puede ser transcripcionalmente controlada por las 3 ramas de la UPR, nuestros resultados indican que esta chaperona podría ser regulada por XBP1s en células cardíacas humanas, pues sus niveles muestran una elevada correlación en nuestro modelo.
La dieta tipo Western, debido a su elevado contenido en grasas, favorece el desarrollo de la resistencia a la insulina y la diabetes de tipo 2, de la misma manera que induce la activación del estrés del RE y la inflamación31,37. Nuestros estudios realizados in vivo demuestran cómo la alimentación de ratones con dietas ricas en ácidos grasos saturados induce un incremento de la expresión de BiP/GRP78 y CHOP en el corazón. No obstante, y al contrario de lo que sucedía en células cardíacas humanas, el corazón de los ratones alimentados con la dieta HFD mostró un incremento en la fosforilación de eIF2α. Algunos de los genes que escapan del control traduccional de eIF2α cuando este es fosforilado son ATF4 y la fosfatasa GADD34. Puesto que GADD34 es capaz de defosforilar eIF2α al cabo de solo 6 h después de producirse el estímulo, es posible que esta actividad fosfatasa sea la responsable de la ausencia de cambios en la fosforilación de eIF2α observada in vitro en nuestro modelo. Aunque no se pueden descartar diferencias entre especies, es factible que esto no suceda en corazón de ratón debido a la continua exposición a los ácidos grasos saturados que se produce in vivo.
Aunque el corazón no es un órgano principal para el almacenamiento de lípidos, sí que puede acumular ácidos grasos, fosfolípidos y triglicéridos en los cardiomiocitos, particularmente cuando la disponibilidad de estos lípidos se encuentra incrementada, tal y como sucede durante la obesidad y la diabetes38. Si esta acumulación persiste en el tiempo, el corazón comenzará a acumular intermediarios lipídicos tóxicos, los cuales están ligados al posterior desarrollo de la resistencia a la insulina y la cardiomiopatía lipotóxica, derivando finalmente hacia la apoptosis de los miocitos y la disfunción contráctil39. Aunque los mecanismos precisos mediante los cuales los ácidos grasos saturados o las dietas ricas en lípidos provocan la apoptosis de los cardiomiocitos no han sido completamente elucidados, diversas evidencias señalan el estrés del RE como una de las causas principales32,35. Así, un estrés del RE prolongado o severo, como por ejemplo el que se produce durante la cardiomiopatía diabética, provoca la muerte por apoptosis de los cardiomiocitos18. Si tenemos en cuenta que los cardiomiocitos raramente pueden proliferar en el corazón adulto, la pérdida de estos puede llegar a comprometer la función cardíaca. En este sentido, las 3 ramas de la UPR pueden promover la expresión de CHOP, un factor de transcripción con una actividad proapoptótica importante, y que lleva a cabo un papel clave en la muerte de las células cardíacas causada por un estrés del RE crónico durante la insuficiencia cardíaca15 o la hipertrofia cardíaca40. Nuestros estudios han demostrado cómo el tratamiento con Wy-14,643 prevenía el incremento de CHOP inducido por palmitato, de manera similar a como lo hacía la activación de AMPK mediante AICAR. Estos resultados indican que la activación de PPARα con Wy-14,643 podría desempeñar un papel antiapoptótico importante por medio de la inhibición del estrés del RE y abre una nueva vía de estudio para la cardiomiopatía diabética. De acuerdo con nuestros resultados, se ha descrito que el palmitato es capaz de inducir la apoptosis por medio de la activación del estrés del RE41,42, mientras que la activación de PPARβ/δ previene la apoptosis inducida por palmitato en células β-pancreáticas35. Este último estudio indicó además que los efectos antiapoptóticos de la activación de PPARβ/δ eran debidos al aumento de la β-oxidación de los ácidos grasos.
Debido a su papel causativo en enfermedades cardiovasculares asociadas a enfermedades metabólicas como la obesidad y la diabetes, el estrés del RE ha sido postulado como una potencial diana terapéutica. Con este objetivo, se ha examinado el efecto de distintas chaperonas sintéticas para prevenir este estrés, debido a su capacidad para estabilizar proteínas y asistir a su correcto plegamiento dentro del RE, de manera similar a como lo hacen las chaperonas endógenas43. Es el caso, por ejemplo, del ácido 4-fenilbutírico y del ácido tauroursodeoxicólico, que han mostrado efectos beneficiosos sobe la resistencia a la insulina, la obesidad y la diabetes en distintos modelos in vitro8,9 e in vivo44. A pesar de ello, estas chaperonas químicas presentan 2 grandes inconvenientes: su baja especificidad y la necesidad de dosis elevadas para conseguir ser eficaces. Es por ello que la investigación y el desarrollo de nuevos fármacos que inhiban el estrés del RE durante las enfermedades metabólicas han despertado un gran interés. La activación de AMPK produce múltiples efectos protectores, incluyendo la inhibición de la inflamación, el estrés oxidativo y la resistencia a la insulina, que resultan en un menor riesgo de obesidad y diabetes de tipo 245. Puesto que la inhibición de la AMPK promueve el estrés del RE, se ha indicado que esta cinasa podría actuar como represora fisiológica de este estrés. En este sentido, recientemente se han publicado distintos trabajos donde se demuestra que la AMPK protege de la lesión isquémica del miocardio, la hipertrofia cardíaca y la aterosclerosis por medio de la inhibición del estrés del RE29,46. De manera similar, se ha descrito que la activación de la AMPK con AICAR previene el estrés del ER en células musculares esqueléticas tratadas con palmitato8. Cuando nosotros examinamos los mecanismos potencialmente responsables del incremento del estrés después de la exposición a palmitato, observamos cómo este ácido graso saturado reducía la actividad AMPK determinada por una disminución de los niveles de fosfo-ACC2. ACC2 es una de las proteínas diana de la cinasa AMPK cuya fosforilación en Ser218 inhibe su actividad. En el corazón, ACC2 es responsable de la conversión de acetil-CoA a malonil-CoA, un conocido inhibidor de la CPT-1, la enzima mitocondrial que interviene en el transporte de ácidos grasos de cadena larga a través de la membrana para su subsiguiente β-oxidación. Por lo tanto, se puede inferir de estos datos que el aumento de la fosforilación de AMPK observado después del tratamiento con palmitato se debe a que la célula trata de compensar la reducción de la actividad ACC2 y, con ello, de la capacidad de oxidación de ácidos grasos. En consonancia con todo esto, el cotratamiento con AICAR previno el aumento de la mayoría de los marcadores de estrés en cardiomiocitos humanos. Resultados similares fueron obtenidos después de inducir el estrés del RE mediante tunicamicina. Más importante aún, nuestros experimentos indican que los efectos preventivos que tiene Wy-14,643 sobre el estrés del RE inducido por palmitato son dependientes de la activación de esta cinasa. Esto se demuestra por los datos obtenidos después de la coincubación de nuestras células con el inhibidor de la AMPK, el Compuesto C, y también por el incremento de la fosforilación de la AMPK y la ACC2 en presencia del agonista de PPARα. Igualmente, los resultados obtenidos en ratón indican que la dieta HFD reduce la β-oxidación de ácidos grasos en corazón. Estos resultados concuerdan con aquellos obtenidos en hígado y músculo de ratón alimentado con una dieta HFD, donde los ácidos grasos saturados parecen contribuir a la inhibición de la AMPK47. En conjunto, nuestros resultados están en la misma línea que los descritos en células β-pancreáticas e hígado48, donde se reportó que la activación de la oxidación de los ácidos grasos por parte de agonistas de PPAR era capaz de prevenir el estrés del RE. El hecho de que el efecto de Wy-14,643 solo se produjera sobre el estrés del RE inducido por palmitato, pero no por tunicamicina, podría explicarse porque esta induce un estrés más severo o, más probablemente, por la presencia de mecanismos adicionales de inducción del estrés del RE por parte de la tunicamicina.
En resumen, los resultados aquí presentados demuestran que la activación de PPARα con Wy-14,643 atenúa el estrés del RE inducido por palmitato en células cardíacas humanas por medio de la activación de AMPK. PPARα regula muchas funciones fisiológicas valiosas que van desde el aumento del catabolismo de los ácidos grasos y la mejora de la sensibilidad a la insulina, hasta la inhibición de la inflamación, mostrando de este modo un papel terapéutico potencial para la prevención y el tratamiento de enfermedades como la diabetes, las dislipidemias o el síndrome metabólico. Dado que la inflamación crónica de baja intensidad y el estrés del RE desempeñan un papel importante en la hipertrofia cardíaca y la insuficiencia cardíaca, y que los agonistas de PPARα se han demostrado eficaces para mejorar e incluso prevenir las enfermedades cardiovasculares asociadas a la obesidad y la diabetes, es tentador especular que este receptor nuclear podría ser una diana terapéutica útil para prevenir la hipertrofia cardíaca y la insuficiencia cardíaca inducidas por el estrés del RE durante estos trastornos metabólicos.
FinanciaciónEste estudio ha sido financiado por fondos del Ministerio de Economía y Competitividad del Gobierno español (SAF2009-06939 y SAF2012-30708). CIBER de Diabetes y Enfermedades Metabólicas Asociadas (CIBERDEM) es una iniciativa del Instituto de Salud Carlos III (ISCIII), Ministerio de Economía y Competitividad.
AutoríaXP contribuyó en el diseño del estudio, la recogida e interpretación de los datos, y en la redacción del artículo. ECB, GG y MMD contribuyeron en el diseño, la interpretación de los datos, y la revisión del artículo. MVC contribuyó en el diseño del estudio, así como en la revisión y aprobación del artículo.
Responsabilidades éticasProtección de personas y animalesLos autores declaran que los procedimientos seguidos se conformaron a las normas éticas del comité de experimentación humana responsable y de acuerdo con la Asociación Médica Mundial y la Declaración de Helsinki.
Confidencialidad de los datosLos autores declaran que en este artículo no aparecen datos de pacientes.
Derecho a la privacidad y consentimiento informadoLos autores han obtenido el consentimiento informado de los pacientes y/o sujetos referidos en el artículo. Este documento obra en poder del autor de correspondencia.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
Una comunicación referente a esta línea de trabajo, titulada «La activación de AMPK previene el estrés del retículo endoplasmático inducido por palmitato en cardiomiocitos humanos AC16», fue presentada en el XXV Congreso Nacional de la SEA-Reus 2012 y galardonada con una Mención especial.