








La quilomicronemia familiar (QF) es un trastorno muy infrecuente, infradiagnosticado y que puede provocar dolor abdominal y pancreatitis recurrente desde la infancia —potencialmente amenazan la vida—, y complicaciones crónicas como diabetes mellitus e insuficiencia pancreática exocrina. La QF afecta a la calidad de vida y salud mental de quienes la padecen, aspectos que hay que tener en cuenta en su tratamiento, basado en una dieta estricta baja en grasas, de difícil adherencia y persistencia. Las personas con QF carecen de capacidad lipolítica para hidrolizar los triglicéridos (TG) y tienen una respuesta mínima o nula a los tratamientos hipolipemiantes convencionales. Los antagonistas de la apoCIII, específicamente volanesorsen, olezarsen y ARO-APOC3, son los fármacos más prometedores para reducir las concentraciones de TG en los pacientes con QF. Las terapias anti-ANGPTL3 parecen ser menos efectivas. Son necesarios más ensayos clínicos y nuevos tratamientos farmacológicos, para mejorar la calidad de vida y el pronóstico de las personas con QF.
Familial chylomicronemia syndrome (FCS) is a very rare, underdiagnosed disorder that can cause abdominal pain and recurrent pancreatitis from childhood —potentially life-threatening— and chronic complications such as diabetes mellitus and exocrine pancreatic insufficiency. FCS affects the quality of life and mental health of those who suffer from it, aspects that must be taken into account in its treatment, based on a strict low-fat diet, which is difficult to adhere to and persist. People with FCS lack the lipolytic capacity to hydrolyze triglycerides (TG) and have a minimal or null response to conventional lipid-lowering treatments. ApoCIII antagonists, specifically volanesorsen, olezarsen and ARO-APOC3, are the most promising drugs to reduce TG concentrations in patients with FCS. Anti-ANGPTL3 therapies appear to be less effective. More clinical trials and new pharmacological treatments are needed to improve the quality of life and prognosis of people with FCS.
La QF se caracteriza clínicamente, además de HTG grave, por episodios recurrentes de pancreatitis desde la infancia, ocasional presencia de xantomas, lipemiaretinalis y un importante impacto sobre la calidad de vida y la salud mental de las personas afectadas1,2. Una de las características más importantes de esta enfermedad es el retraso en el diagnóstico, por tratarse de una enfermedad poco frecuente y, por tanto, desconocida por la mayoría de los profesionales sanitarios. De hecho, según datos iniciales del estudio APPROACH3, la edad media en el momento del diagnóstico fue de 24 años.
PrevalenciaSe trata de un trastorno muy poco frecuente y de prevalencia desconocida, en parte por la dificultad para diagnosticarla ya que el estudio genético no está siempre disponible. En Europa y según diferentes estudios, afecta aproximadamente entre una y 19 personas por millón de personas4–6. En nuestro país existen al menos 2 registros7,8 de 13 y 26 pacientes con genética compatible.
FisiopatologíaEl trastorno fundamental es la ausencia de actividad prácticamente total de la enzima LPL, enzima clave en la hidrólisis y catabolismo de los TG, debido a la existencia de variantes patogénicas bialélicas con pérdida de función en los 5 genes previamente detallados (LPL, GPIHBP1, APOA5, APOC2 y LMF1)9.
En condiciones normales los quilomicrones (Qm) son sintetizados en el intestino tras la ingesta de grasa, y transportan grandes cantidades de TG (vía exógena) que son hidrolizados por la LPL, que precisa para su normal funcionamiento de otras proteínas que actúan como cofactores, como la ApoAV, la ApoCII, la LMF1 y la GPIHBP1. El resto de Qm son absorbidos por el hígado a través de los receptores de LDL y el denominado receptor LRP (proteínas relacionadas con el receptor LDL) que utilizan la apolipoproteína E (ApoE) como ligando10. Los Qm desaparecen del plasma a las 4-6h, por lo que en condiciones fisiológicas no son detectados en ayunas (fig. 1). Lo que caracteriza a este trastorno es la persistencia de los Qm en plasma, incluso en ayunas de más de 14h debido a la imposibilidad de ser metabolizados ya que al no existir prácticamente actividad de la LPL no pueden ser metabolizados.
Manifestaciones clínicas
El contenido de la grasa de la dieta es fundamental para evitarlas principales manifestaciones; es decir, episodios de pancreatitis y dolor abdominal (fig. 2). La pancreatitis aguda recurrente11 es una complicación grave que pone en peligro la vida del paciente y que puede producir complicaciones a largo plazo como insuficiencia pancreática exocrina, necrosis parcial o total de la glándula pancreática y diabetes secundaria. Existe, además, una correlación entre las concentraciones de TG y el desarrollo de pancreatitis. Es más, aunque habitualmente las pancreatitis agudas suelen suceder con cifras muy elevadas de TG, por encima de 2.000mg/dl, cifras mucho menores también pueden desencadenar una pancreatitis12,13. Un aumento de 100mg/dl de las concentraciones de TG, incrementa el riesgo de producir una pancreatitis aguda aproximadamente en un 4%. No se conocen con exactitud los mecanismos fisiopatológicos por los que la quilomicronemia produce la pancreatitis. Se ha postulado que la hiperviscosidad sanguínea secundaria al exceso de Qm circulantes provocaría isquemia local y acidosis pancreática. Otros mecanismos incluirían el exceso de radicales oxidados provenientes de los ácidos grasos libres mal hidrolizados por la lipasa pancreática o la inflamación mitocondrial de las células pancreáticas1. No hay correlación entre el valor máximo absoluto de TG y la gravedad de la pancreatitis14.

Los pacientes con QF suelen tener episodios recurrentes de dolor abdominal con o sin pancreatitis, provocando el ayuno voluntario en muchas ocasiones. Otras manifestaciones incluyen la aparición de xantomas eruptivos, náuseas, vómitos, lipemia retinalis, hepatoesplenomegalia, retraso en el crecimiento, cuadros confusionales con «niebla mental» y alteraciones en el estado de ánimo y de la salud mental. La prevalencia de los distintos síntomas varía según resultados publicados de distintas cohortes8,15,16.
Un aspecto muy relevante es el impacto sobre la calidad de vida de estos pacientes, experimentando a menudo miedo y ansiedad a presentar nuevos episodios de pancreatitis16–18. Las relaciones sociales y su mundo afectivo se ven también afectadas19. Los pacientes experimentan frustración ante la incomprensión de los familiares y amigos sobre la naturaleza de su enfermedad y las implicaciones sobre el tratamiento de esta. Hasta el 23% de los pacientes manifiestan trastornos de la conducta alimentaria.
DiagnósticoEl diagnóstico es genético. Ante un paciente con HTG grave (>880mg/dl) y presencia de Qm en la ultracentrifugación, en ausencia de causas secundarias (ingesta de alcohol, diabetes mal controlada, obesidad, resistencia insulínica, fármacos, enfermedad renal crónica, paraproteinemias, etc.) debe valorarse solicitar un estudio genético. Dado que se trata de una enfermedad muy poco frecuente, y la clínica puede pasar desapercibida respecto a otras etiologías, Moulin et al.20, han desarrollado un algoritmo diagnóstico de sospecha de QF. En general una apolipoproteína B (ApoB) normal/baja, junto con un índice de masa corporal normal o bajo, síntomas recurrentes de dolor abdominal desde la infancia, HTG grave en múltiples ocasiones, pancreatitis de repetición y ausencia de respuesta al tratamiento hipolipemiante convencional, hacen más plausible el diagnóstico de QF y menos probable el de SQM.
Cuando el diagnóstico genético no esté disponible, o sea negativo, a pesar de una alta sospecha clínica, podría medirse la actividad de la LPL11 tras inyección de heparina. Una actividad de LPL menor al 20% confirmaría el diagnóstico, aunque lamentablemente esta técnica solo está disponible en centros de investigación y no se realiza de rutina.
Recientemente se han descrito la presencia de autoanticuerpos21,22 dirigidos contra la LPL o contra la proteína GPIHBP1. Por la tanto, la posibilidad de una quilomicronemia autoinmune habría que considerarlo cuando la genética sea negativa. Desafortunadamente, el test que detecta la presencia de autoanticuerpos no está disponible en la mayoría de los centros.
TratamientoEl tratamiento debe de comprender un abordaje global del paciente, que incluya, como objetivos prioritarios evitar episodios de pancreatitis recurrentes, controlar la HTG crónica y por ende la quilomicronemia, y tratar y abordar la salud mental y mejorar en lo posible la calidad de vida de los pacientes. Los pilares fundamentales del tratamiento son la dieta y el tratamiento farmacológico. La dieta debe de ser muy restrictiva en grasas23,24 (<20 g al día de cualquier tipo de grasa), por lo que es necesario la participación de dietistas y nutricionistas en el manejo de los pacientes. La fuente principal de energía serán los hidratos de carbono y los triglicéridos de cadena media (TCM), que se pueden utilizar como aceite en crudo. En la tabla 1 hay un resumen de los alimentos a evitar y de los que pueden ingerir sin problema.
Recomendaciones sobre grupos de alimentos en el síndrome de quilomicronemia familiar
| Alimentos a incluir en la dieta | Alimentos a evitar en la dieta |
|---|---|
| Verduras y legumbres sin aceite añadido | Alcohol |
| Cereales integrales | Azúcares añadidos |
| Leche desnatada | Bebidas azucaradas y zumos |
| Carne magra | Pastelería y bollería industrial |
| Productos desnatados | Aceites de cualquier tipo |
| Hidratos de carbono complejos | Alimentos procesados/ultraprocesados |
| Ácidos grasos esenciales como linoleico y α-linoleico | Alimentos con grasas |
En los episodios de pancreatitis aguda, el manejo de estos pacientes es el convencional del resto de etiologías que puedan provocar una pancreatitis. Así, el reposo digestivo con dieta absoluta, reposición de hidroelectrolíticos con fluidoterapia endovenosa y analgesia suelen ser eficaces para disminuir rápidamente la HTG y controlar el dolor. Las terapias de recambio plasmático no han demostrado superioridad a largo plazo sobre el tratamiento convencional quedando limitado su uso en caso de embarazo. En los pacientes diabéticos con hiperglucemia, la insulina endovenosa puede ser útil.
Tratamiento farmacológico del SQF: volanesorsenEl tratamiento farmacológico es imperativo para controlar la quilomicronemia en la QF, pues la implementación de recomendaciones de medidas alimentarias, aun siendo necesarias, no son suficientes. En ese sentido, los fibratos pueden contribuir a disminuir la sobreproducción hepática de VLDL y mejorar los niveles de TG cuando existe un fenotipo V, pero no corrigen la quilomicronemia en sujetos con QF.
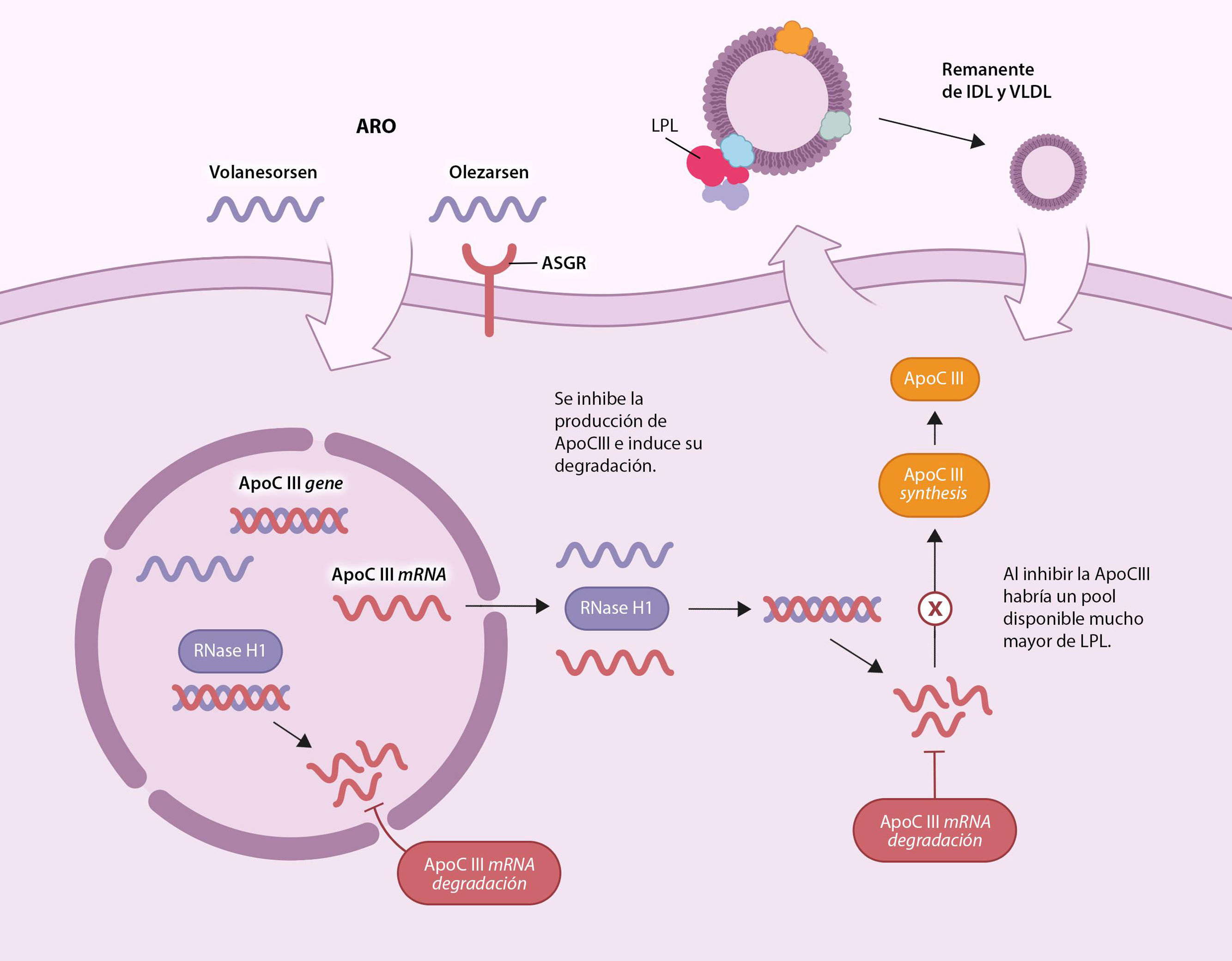
Así, volanesorsen es el único fármaco aprobado por la Agencia Europea del Medicamento (EMA, por sus siglas en inglés) y disponible en nuestro país, con eficacia demostrada en QF. Se trata de un oligonucleótido antisentido que en el núcleo de la célula hibrida al RNA mensajero de la apolipoproteína CIII (ASO-ApoCIII) y fuerza a la célula a su destrucción (fig. 3). Queda por aclarar el mecanismo de acción último por el que reduce la HTG, aunque en este sentido se ha postulado que al disminuir ApoCIII (Apo-CIII) habría un pool disponible mucho mayor de LPL, si bien en estos pacientes la LPL no recupera la actividad. El estudio pivotal de volanesorsen fue el ensayo APPROACH25, un ensayo clínico de fase 3 que incluyó 66 pacientes con SQF aleatorizados 1:1 a recibir volanesorsen 285mg o placebo, ambos de administración subcutánea y semanal durante 52 semanas. A los 3 meses, el descenso en la concentración de TG (objetivo primario) fue del 77% que corresponde a una reducción de TG de 1.712mg/dl, mientras la concentración de Apo-CIII se redujo en un 84%. Finalmente, un 77% de los participantes tratados con volanesorsen, alcanzaron concentraciones medias de TG por debajo de 750mg/dl versus 18% en el grupo placebo. Como efectos adversos más importantes destacaron las reacciones locales en el lugar de la inyección (enrojecimiento y prurito) y trombocitopenia por debajo de 100.000 plaquetas/mm3 en 15 de los 33 pacientes tratados con volanesorsen.

En España las condiciones de financiación para la utilización de volanesorsen incluyen el diagnóstico genético confirmado de QF (variante bialélica), pancreatitis de repetición y concentraciones de TG superiores a 750mg/dl. Los resultados clínicos en el seguimiento de pacientes tratados con volanesorsen deben ser incluidos en la plataforma VALTERMED del Ministerio de Sanidad, siendo necesario revisar la efectividad del tratamiento a los 3 meses, discontinuándolo si no se consigue una reducción de TG superior al 25% sobre el nivel basal y niveles por debajo de 750mg/dl (8,5mmol/l) o si dicha concentración no baja de 2.000mg/dl (22,6mmol/l, tras 3 meses de tratamiento semanal con 285mg. También será necesaria una evaluación periódica, al menos cada 3 meses, retirando el fármaco por inefectividad si los niveles de TG vuelven a aumentar con el tiempo hasta niveles similares a los del inicio del tratamiento.
El efecto secundario que más limita su utilización ha sido la trombocitopenia. Hay que recordar que muchos de estos pacientes han tenido pancreatitis previas y presentan ya complicaciones como esplenomegalia o trombosis de la arteria esplénica, que hacen que su recuento plaquetario en muchas ocasiones sea más bajo de lo normal. En la ficha técnica queda reflejado que no se iniciará el tratamiento en los pacientes con trombocitopenia (plaquetas <140.000/mm3).
Otros tratamientos farmacológicos en desarrolloOlezarsen: oligonucleótido antisentido inhibidor de Apo-CIII (ASO-ApoCIII) conjugado con N-acetilgalactosamina triantenaria (GalNAc3), un ligando de hidratos de carbono para los receptores de asialoglicoproteína que abundan en la superficie de los hepatocitos, facilitando así su entrada en el núcleo de los mismos donde se genera el RNA mensajero de Apo-CIII (fig. 3) e inhibiendo así la producción de Apo-CIII. Recientemente se ha publicado un ensayo clínico26 de fase 3, en el que se incluyeron 66 pacientes con SQF, siendo 22 de ellos asignados al grupo de tratamiento con 80mg de olezarsen, 21 al grupo de 50mg de olezarsen y 23 al grupo de placebo todos ellos administrados subcutáneamente y cada 4 semanas. Al inicio del estudio, el nivel medio (± desviación estándar) de TG fue de 2.630±1.315mg/dl, y el 71% tenía antecedentes de pancreatitis aguda en los 10 años anteriores. Los niveles de TG a los 6 meses se redujeron significativamente con la dosis de 80mg de olezarsen (−43,5%; intervalo de confianza [IC] del 95%: −69,1 a −17,9; p<0,001), pero no con la dosis de 50mg (−22,4%; IC del 95%: −47,2-2,5; p=0,08). La diferencia en el cambio porcentual medio en el nivel de apolipoproteína C-III desde el inicio hasta los 6 meses en el grupo de 80mg en comparación con el grupo de placebo fue del −73,7% (IC del 95%: −94,6 a −52,8) y entre el grupo de 50mg en comparación con el grupo de placebo fue del −65,5% (IC del 95%: −82,6 a −48,3). A las 53 semanas, se habían producido 11 episodios de pancreatitis aguda en el grupo de placebo y un episodio en cada grupo de olezarsen (cociente de tasas grupos combinados de olezarsen versus placebo): 0,12; IC del 95%: 0,02-0,66. No hubo tasas significativas de trombocitopenia.
Plozasiran: ARN pequeño de interferencia (ARO-ApoCIII) de acción hepática que reduce la producción de Apo-CIII. Muy recientemente se ha publicado un ensayo27 de fase 3, doble ciego y aleatorizado, en el que se evaluaba la eficacia y la seguridad de plozasiran, en comparación con placebo, entre adultos con HTG grave (<1.000mg/dl) en al menos 3 análisis consecutivos, resistente a terapia hipolipemiante estándar, y por lo menos otro criterio adicional (diagnóstico genético previo de SQF, actividad de LPL posheparina ausente o baja —inferior al 20% del valor normal— antecedentes de pancreatitis aguda no causada por alcohol o colelitiasis, hospitalizaciones recurrentes por dolor abdominal intenso sin otra causa identificada, pancreatitis infantil o antecedentes familiares de pancreatitis inducida por HTG). Así, 75 pacientes fueron asignados aleatoriamente en una proporción de 2:1:2:1 para recibir 25mg de plozasiran o placebo de volumen equivalente o para recibir 50mg de plozasiran o placebo de volumen equivalente por vía subcutánea cada 3 meses durante 12 meses. A los 10 meses, el cambio medio desde el inicio en las concentraciones de TG en ayunas (criterio de valoración principal) fueron del −80% en el grupo de 25mg de plozasiran, del −78% en el grupo de 50mg de plozasiran y del −17% en el grupo placebo (p<0,001). Los criterios de valoración secundarios mostraron mejores resultados en los grupos de plozasiran que en el grupo placebo, incluida la incidencia de pancreatitis aguda (odds ratio: 0,17; IC del 95%: 0,03-0,94; p=0,03). El riesgo de eventos adversos fue similar en todos los grupos, mientras los eventos adversos graves y serios fueron menos comunes con plozasiran que con placebo. Se produjo hiperglucemia con plozasiran en algunos pacientes con prediabetes o diabetes al inicio del tratamiento. Las concentraciones de TG en los pacientes con quilomicronemia persistente fueron significativamente menores con plozasiran que con placebo, así como la incidencia de pancreatitis aguda.
Evinacumab: la proteína similar a la angiopoyetina 3 (ANGPTL3) es una proteína circulante codificada por el gen ANGPTL3 en el cromosoma 1p31 que se sintetiza en el hígado y regula parcialmente el metabolismo lipídico al inhibir la actividad de la LPL y la lipasa endotelial. El evinacumab28 es un anticuerpo monoclonal dirigido contra la ANGPLT3, aprobado para el tratamiento de la hipercolesterolemia familiar homocigota en función de los resultados del ensayo ELIPSE HoFH29, ensayo en el que los pacientes que recibieron tratamiento con evinacumab, mostraron una reducción media de la concentración de TG en un 55%. Para evaluar el potencial de evinacumab en los pacientes con HTG grave, se realizó un ensayo30 de fase 2, aleatorizado, doble ciego (evinacumab versus placebo) en los pacientes con HTG grave y al menos una hospitalización previa por pancreatitis aguda. Una de las cohortes de este estudio incluyó solo pacientes con SQF; en esa cohorte, evinacumab no se asoció con una reducción en los niveles de TG en comparación con placebo. Quizás se necesiten dosis muy elevadas de evinacumab para los pacientes en los que la actividad de la LPL sea nula o esté muy disminuida. Actualmente está en marcha un ensayo clínico para los pacientes con SQM.
Lomitapida: es un fármaco que disminuye la actividad de la proteína microsomal transferidora de triglicéridos (MTP) lo que consigue reducir las concentraciones de TG al impedir la transferencia de TG a lipoproteínas nacientes que contienen Apo B, incluidos los quilomicrones31. Inicialmente se ha aprobado su uso en la hipercolesterolemia familiar homocigota, habiendo sido utilizado de forma anecdótica en un paciente con SQF con aceptable resultado clínico, pero con empeoramiento notable del hígado graso del paciente32.
FinanciaciónEste trabajo fue financiado por una beca no condicionada de Sobi, que no participó en el diseño del mismo ni en la elaboración de este manuscrito.
Información sobre el suplementoEste artículo forma parte del suplemento titulado « Hipertrigliceridemia severa. De las bases genéticas a la pràctica clínica », que ha sido financiado por la Sociedad Española de Arteriosclerosis, con patrocinio de Sobi.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.