







Durante décadas, se ha considerado a la quilomicronemia (HTG) familiar como una entidad específica caracterizada por un aumento de las partículas de lipoproteínas de muy baja densidad (VLDL) y un patrón de herencia autosómico dominante. En la era de la genómica, se ha comprobado que la HTG familiar, si bien podía agruparse en familias, tenía una herencia poligénica en la que el fenotipo estaría determinado por factores ambientales concomitantes. De ahí su inclusión en el grupo de las HTG poligénicas. Clínicamente, se caracterizan por HTG moderadas, con el consiguiente aumento del riesgo cardiovascular, y en raras ocasiones, por HTG severas con riesgo de pancreatitis agudas. El tratamiento se basará en controlar los factores ambientales, implementar medidas higiénico-dietéticas y, en ocasiones, fármacos, para disminuir el riesgo cardiovascular en las HTG moderadas y de pancreatitis aguda en las HTG severas.
For decades, familial hypertriglyceridemia (HTG) has been considered a specific entity characterized by an increase in VLDL particles and an autosomal dominant inheritance pattern. In the genomics era, it has been proven that familial HTG, although it could be grouped in families, had a polygenic inheritance in which the phenotype would be determined by concomitant environmental factors. Hence its inclusion in the group of polygenic HTGs. Clinically, they are characterized by moderate HTG, with the consequent increase in cardiovascular risk, and in rare cases, by severe HTG with risk of acute pancreatitis. Treatment will be based on controlling environmental factors, implementing hygienic-dietetic measures and sometimes drugs, to reduce cardiovascular risk in moderate HTGs and acute pancreatitis risk in severe HTGs.
La quilomicronemia (HTG) se define como un aumento de la concentración sérica de triglicéridos (TG) en ayunas por encima de 150 mg/dL (> 1,7 mmol/L)1. La HTG se debe a un acúmulo de las lipoproteínas ricas en triglicéridos (LRT) y de sus remanentes, ya sea por aumento de su síntesis, disminución de su aclaramiento o de ambas a la vez. En EEUU, un 16,3% de la población tiene una concentración de TG entre 200 y 500 mg/dL, y un 1,7% por encima de 500 mg/dL2. En nuestro país, en un estudio con más de medio millón de trabajadores, el 16% tenía una HTG leve (150 a 399 mg/dL), un 1,1% moderada (400 a 999 mg/dL) y un 0,03% severa (≥ 1.000 mg/dL)3. La prevalencia de la HTG se ha ido incrementando en los últimos años asociado al aumento de la prevalencia de diabetes y de obesidad.
Clásicamente, las HTG se han clasificado según su etiología en primarias o secundarias. Las primarias se debían fundamentalmente a factores genéticos, mientras que las secundarias eran provocadas por factores ambientales, fármacos u otras enfermedades. Si bien, la mayoría de los pacientes con HTG tienen, al menos, algún factor secundario, no todos los que son portadores de estos desarrollan HTG, lo que sugiere que, en los individuos que desarrollan la HTG, existe, además, una base genética predisponente, y esta es de carácter poligénico. Además, la gravedad individual de una HTG secundaria está probablemente determinada por una susceptibilidad genética.
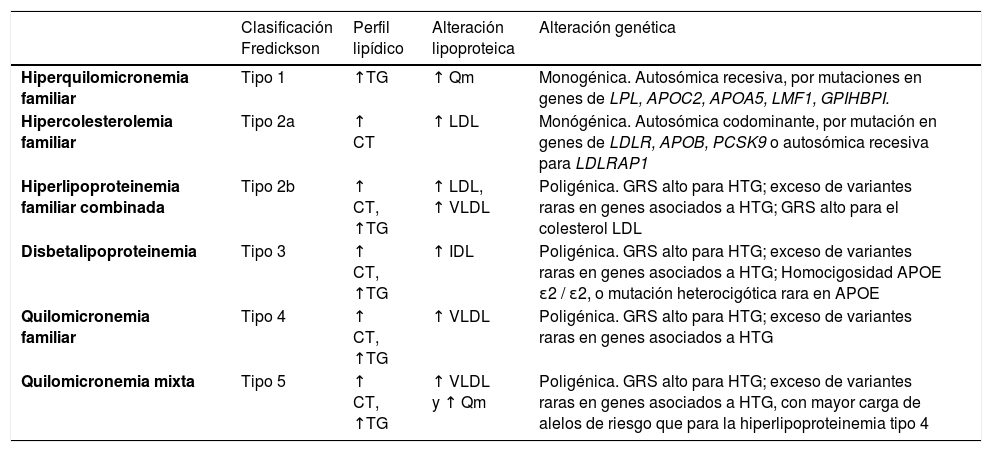
Clasificación de las hiperlipoproteinemias en la era pregenómicaDurante décadas se ha utilizado la clasificación de Fredrickson4 que fue posteriormente incluida en la lista de enfermedades de la Organización Mundial de la Salud (OMS). Esta división de las hiperlipoproteinemias se hizo de acuerdo con su fenotipo, está basada en los patrones electroforéticos de las fracciones lipoproteicas. Se describen seis fenotipos, y en cinco de ellos existe un incremento en la concentración de TG. Los diferentes fenotipos asociados a la HTG se definen por la clase o clases específicas de partículas acumuladas de LRT, incluyendo quilomicrones, lipoproteínas de muy baja densidad (VLDL) y sus remanentes (tabla 1). Este sistema de clasificación se basaba en la idea de que la diferencia entre los fenotipos asociados con las HTG se debía a variaciones genéticas; sin embargo, numerosos datos recientes sugieren que esto no es así5–7. Por ejemplo, la hiperlipoproteinemia tipo IV provocada por un incremento de las partículas de VLDL circulantes correspondería a la HTG familiar, que en un principio, se creía que tenía un patrón de herencia autosómico dominante. Posteriormente, se ha comprobado que, si bien los casos de HTG podían agruparse en familias, la HTG dentro de una familia generalmente no seguía patrones mendelianos clásicos de herencia y no mostraba una transmisión vertical constante entre generaciones, lo cual correspondería con una herencia tipo poligénica, en la que el fenotipo clínico estaría determinado en gran medida por factores ambientales concomitantes8.
Clasificación clásica de las hiperlipoproteinemias de Fredrickson según el fenotipo lipídico
| Clasificación Fredickson | Perfil lipídico | Alteración lipoproteica | Alteración genética | |
|---|---|---|---|---|
| Hiperquilomicronemia familiar | Tipo 1 | ↑TG | ↑ Qm | Monogénica. Autosómica recesiva, por mutaciones en genes de LPL, APOC2, APOA5, LMF1, GPIHBPI. |
| Hipercolesterolemia familiar | Tipo 2a | ↑ CT | ↑ LDL | Monógénica. Autosómica codominante, por mutación en genes de LDLR, APOB, PCSK9 o autosómica recesiva para LDLRAP1 |
| Hiperlipoproteinemia familiar combinada | Tipo 2b | ↑ CT, ↑TG | ↑ LDL, ↑ VLDL | Poligénica. GRS alto para HTG; exceso de variantes raras en genes asociados a HTG; GRS alto para el colesterol LDL |
| Disbetalipoproteinemia | Tipo 3 | ↑ CT, ↑TG | ↑ IDL | Poligénica. GRS alto para HTG; exceso de variantes raras en genes asociados a HTG; Homocigosidad APOE ɛ2 / ɛ2, o mutación heterocigótica rara en APOE |
| Quilomicronemia familiar | Tipo 4 | ↑ CT, ↑TG | ↑ VLDL | Poligénica. GRS alto para HTG; exceso de variantes raras en genes asociados a HTG |
| Quilomicronemia mixta | Tipo 5 | ↑ CT, ↑TG | ↑ VLDL y ↑ Qm | Poligénica. GRS alto para HTG; exceso de variantes raras en genes asociados a HTG, con mayor carga de alelos de riesgo que para la hiperlipoproteinemia tipo 4 |
APOE: apolipoproteína E; CT: colesterol total; GRS: puntuación de riesgo poligénico; HTG: quilomicronemia; IDL: lipoproteínas de densidad intermedia; LDL: lipoproteínas de baja densidad; Qm: quilomicrones; TG: triglicéridos; VLDL: lipoproteínas de muy baja densidad.
Fuente: adaptada de Hegele et al.9
Desafortunadamente, esta clasificación no sirve para el diagnóstico etiológico, no tiene utilidad clínica, no es capaz de predecir los eventos cardiovasculares ni el riesgo de pancreatitis, no informa sobre el pronóstico y tampoco tiene utilidad terapéutica, no diferenciando qué paciente se va a beneficiar más con un tratamiento determinado7,9.
Clasificación de las quilomicronemias en la era de la genómicaEn 2014, el International Consensus Panel on Hypertriglyceridemia9 recomendó evitar el término «familiar» asociado con la HTG, ya que en la mayoría se debe a defectos poligénicos, en lugar de monogénicos y el fenotipo clínico está determinado en gran medida por factores ambientales concomitantes. De acuerdo con esto, se clasifica a las HTG en dos grandes grupos: monogénicas y poligénicas.
Los sujetos con HTG monogénicas son homocigotos o heterocigotos compuestos para una mutación por pérdida de función en los genes LPL, APOC2, LMF1, APOA5 y GPIHBP1. Son enfermedades raras, < una por millón de habitantes, con concentración de TG >1.000 mg/dL, que suelen debutar en la infancia o juventud y que clínicamente tienen un alto riesgo de pancreatitis10.
Las HTG poligénicas son responsables de más del 99% de las HTG. Son generalmente moderadas, con TG entre 2 a 10 mmol/L (177 a 885 mg/dL). Este grupo incluirá, entre otras, la anteriormente denominada HTG familiar. Son el resultado del efecto acumulado de variantes tanto comunes como raras en > 45 loci relacionados con el metabolismo de los TG (tabla 2) con múltiples efectos sumativos sobre la concentración de TG, pero sin la influencia de un único gen11. Estas variaciones genéticas interactúan con factores secundarios no genéticos (tabla 3) dando lugar a un determinado fenotipo lipídico12. En ocasiones, en el curso de una HTG moderada, a expensas de una elevación de VLDL, al añadirse alguno de los factores ambientales mencionados, puede producirse una saturación del sistema de lipólisis intravascular, impidiendo el catabolismo de los quilomicrones, creando una situación, generalmente transitoria, de hiperquilomicronemia secundaria o poligénica13.
Listado de los 45 loci asociados independientemente con los niveles de triglicéridos plasmáticos de forma aislada o en conjunción con otros lípidos plasmáticos
| Lípidos afectados | Loci |
|---|---|
| Solo triglicéridos | MET, AKR1C4, PDXDC1, MPP3, INSR, MSL2L1, KLHL8, MAP3K1, TYW1B, PINX1, JMJD1C, CYP26A1, CAPN3, FRMD5, CTF1, PLA2G6 |
| Triglicéridos y c-HDL | RSPO3, FTO, VEGFA, PEPD, GALNT2, IRS1, PLTP, MLXIPL, LPL, LRP1 |
| Triglicéridos, colesterol total, c-HDL y c-LDL | CETP, TRIB1, FADS1–2–3, APOA5 |
| Triglicéridos, colesterol total y c-LDL | ANGPTL3, MIR148A, LRAP1, TIMD4, CILP2 |
| Triglicéridos, c-HDL y c-LDL | PIGV-NR0B2 |
| Triglicéridos y colesterol total | GCKR, NAT2 |
| Triglicéridos, colesterol total y c-HDL | LIPC |
c-HDL: colesterol unido a lipoproteínas de alta densidad; c-LDL: colesterol unido a lipoproteínas de baja densidad.
Fuente: Willer et al.11
Factores que pueden contribuir a una quilomicronemia en personas con predisposición genética
| Dieta rica en grasa o con índice glicémico elevado |
| Consumo de alcohol |
| Obesidad |
| Diabetes no diagnosticada o mal controlada |
| Síndrome metabólico |
| Hipotiroidismo |
| Embarazo |
| Enfermedad renal (proteinuria, glomerulonefritis y uremia) |
| Paraproteinemia |
| Lupus eritematoso sistémico |
| Fármacos: betabloqueantes no selectivos, diuréticos tiazídicos y del asa, estrógenos, glucocorticoides, antipsicóticos atípicos, ciclosporina, inhibidores de la proteasa, interferón-alfa, sirolimus, tacrolimus, ácido cis-retinoico, moduladores selectivos del receptor estrogénico |
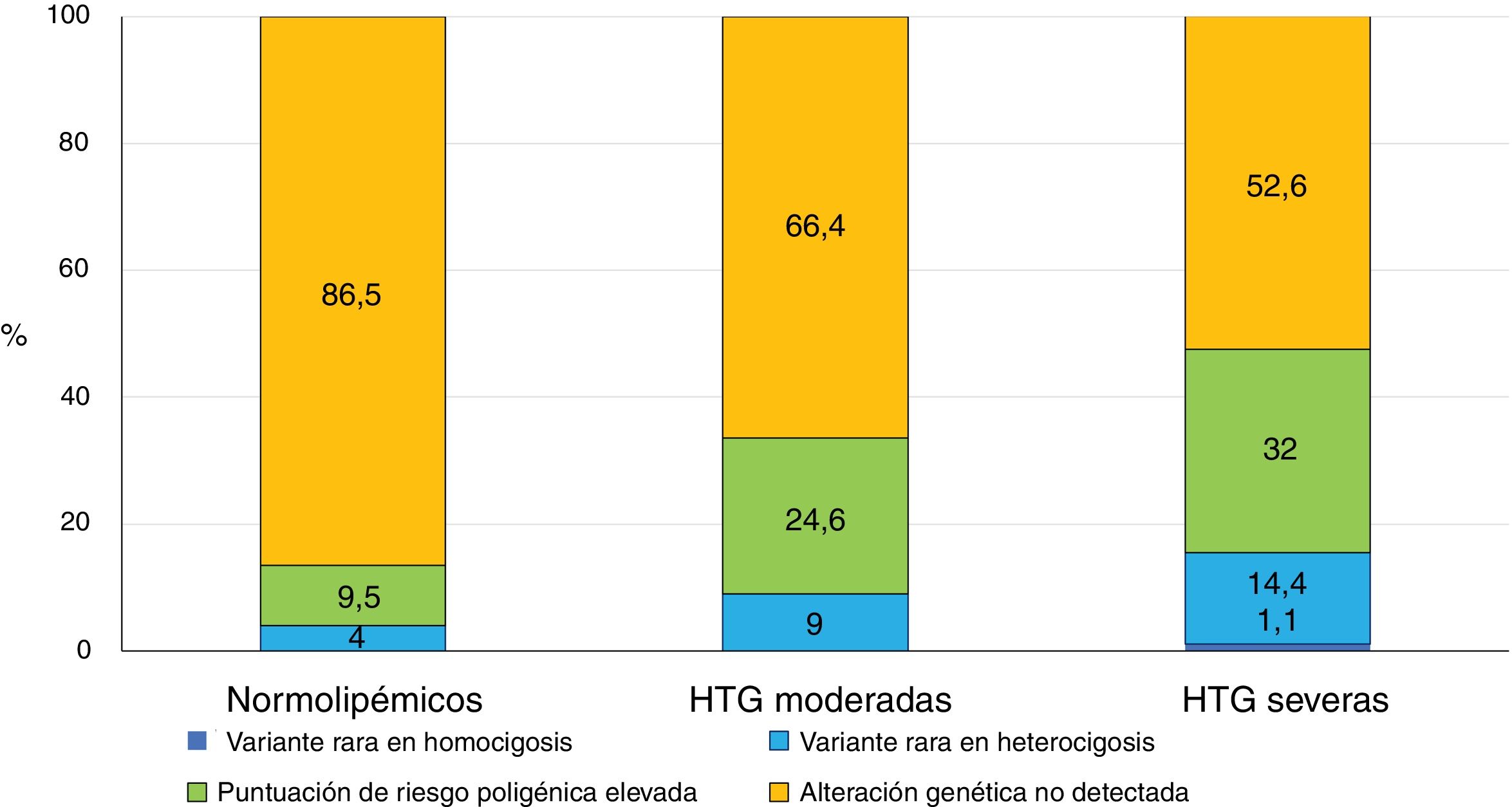
Dron et al.14 estudiaron el perfil genético en 134 pacientes con HTG moderada y lo compararon con el de 563 individuos con HTG severa (>10 mmol/L) y 503 sujetos normolipémicos. Buscaron variantes raras (frecuencia alélica en la población menor del <1%) en los principales genes del metabolismo de los TG (LPL, APOC2, GPIHBP1, APOA5 y LMF1) y estudiaron 16 polimorfismos de nucleótido único (SNPs) asociados con la HTG, con los que desarrollaron una puntuación de riesgo de HTG poligénica15. En más de la mitad de los pacientes con HTG severa no se encontró ninguna alteración genética que la justificara. La prevalencia de variantes raras osciló entre un 4% en población normolipémica, un 9% en HTG moderadas y hasta un 14,4% en HTG severas (fig. 1). El porcentaje de sujetos con una puntuación de riesgo genético de HTG elevada se incrementaba desde un 9,5% en la población general, un 24,6% en HTG moderadas y un 32% en sujetos con HTG severas14. Estos hallazgos confirman que, a mayor gravedad de la HTG, existe una prevalencia más elevada de determinantes genéticos, ya sean variantes raras que alteran los genes del metabolismo de los TG o una acumulación excesiva de SNPs, siendo estos últimos el contribuyente genético más prevalente de las HTG, independientemente de la gravedad de las mismas14. Queda por identificar más de la mitad del componente genético de las HTG que podría provenir de interacciones gen por gen, efectos epigenéticos u otros efectos no mendelianos, o la influencia de interacciones del gen con el medio ambiente8.

Comparación entre los perfiles genéticos de 3 cohortes de sujetos, normolipémicos, pacientes con quilomicronemia moderada y pacientes con quilomicronemia severa.
Fuente: adaptada de Dron14.
Los estudios genéticos por sí solos no sirven para predecir la gravedad de la expresión fenotípica de la HTG en un paciente concreto. Además, no tenemos evidencias de que los resultados clínicos, el pronóstico o las intervenciones terapéuticas difieran según el genotipo. Por todo ello, actualmente no se recomienda efectuar pruebas genéticas de rutina en individuos con concentraciones de TG de 2 a 10 mmol/L, y solo se recomienda su realización en HTG graves en pacientes pediátricos y en adolescentes8.
Manifestaciones clínicas de las quilomicronemias poligénicasLas HTG poligénicas se caracterizan por elevaciones moderadas de la concentración de TG, en la mayoría de los casos, asociadas con una disminución de la concentración de colesterol-HDL, y concentraciones variables de colesterol unido a lipoproteínas de baja densidad (LDL). La importancia clínica de las HTG reside en su asociación con la enfermedad cardiovascular, con elevaciones moderadas y con la pancreatitis aguda, con elevaciones importantes16.
La HTG, como biomarcador de las lipoproteínas ricas en TG circulantes y de sus remanentes, se ha asociado con el riesgo cardiovascular. Las partículas remanentes, y no las grandes lipoproteínas ricas en TG, promueven la aterogénesis tras su infiltración en la pared del vaso, mediante su actividad proinflamatoria y protrombótica9. Grandes estudios de cohortes han encontrado una asociación entre la concentración de TG con un incremento de la incidencia de cardiopatía isquémica, ictus y mortalidad cardiovascular, tanto en varones como en mujeres17,18. En un metaanálisis de 68 estudios prospectivos con más de 300.000 participantes, el aumento de la concentración de TG se vinculaba con un incremento significativo del riesgo de enfermedad cardiovascular tras ajustar por los factores de riesgo clásicos, pero perdía la significación al ajustar por el colesterol-HDL y por el colesterol-noHDL19. Por otra parte, estudios de aleatorización mendeliana sugieren que las altas concentraciones plasmáticas de lipoproteínas ricas en TG y sus remanentes a lo largo de la vida se relacionan con un mayor riesgo de cardiopatía isquémica, independientemente de las concentraciones de colesterol-HDL20. Sin embargo, ensayos clínicos con fármacos que reducen los TG, pero que también afectan a otros componentes lipídicos, no han demostrado claramente una reducción de los eventos cardiovasculares, si bien tienden a disminuirlos en los sujetos con TG elevados y colesterol-HDL bajo21. En resumen, la HTG en ayunas o posprandial es un marcador de riesgo cardiovascular que podría contribuir a explicar, en parte, el denominado riesgo residual tras la reducción del colesterol-LDL. Hasta el momento, no se ha confirmado de forma definitiva su papel causal, ya que podría formar parte de un fenotipo lipídico más complejo o ser la expresión de un trastorno metabólico subyacente, que fueran los verdaderos responsables del aumento de riesgo22.
La HTG es la tercera causa más frecuente de pancreatitis aguda, tras el consumo de alcohol y la colelitiasis, si bien es la que cursa con un peor pronóstico de las tres. La HGT grave es responsable del 5 al 20% de las pancreatitis agudas23. La pancreatitis suele aparecer en HTG muy importantes en el contexto de un síndrome de hiperquilomicronemia familiar, si bien, también se ha descrito asociada a HTG moderada, no existiendo correlación entre la concentración de TG y el riesgo de pancreatitis aguda24, pero si con la gravedad de la misma25.
Directrices en el manejo de las quilomicronemias poligénicasEn todo paciente con HTG habrá que evitar o corregir los factores exógenos que pueden descontrolar la concentración de TG (tabla 3), como puede ser regular la glucemia en el diabético descontrolado, perder peso en el obeso, frenar el consumo de alcohol o retirar un fármaco que aumenta los TG16. En los individuos con HTG poligénica y concentraciones moderadas de TG el objetivo del tratamiento será reducir el riesgo cardiovascular. Se deberá estimar el riesgo cardiovascular absoluto y cumplir objetivos de colesterol-LDL y colesterol-no HDL. Si una vez alcanzadas las metas persisten elevados los TG habrá que insistir en medidas higiénico-dietéticas, y si no es suficiente, valorar la utilización de fibratos (fenofibrato) o ácidos grasos omega-326. En los raros casos de HTG poligénica y TG severamente elevados, el objetivo del tratamiento será disminuir el riesgo de pancreatitis aguda. Para ello, junto con las medidas higiénico-dietéticas, habrá que iniciar terapia farmacológica, aunque, hasta ahora no está comprobado que esta disminuya el riesgo de desarrollar pancreatitis16.
FinanciaciónEste artículo ha sido financiado con una ayuda sin restricciones por Akcea Therapeutics. El patrocinador no ha intervenido en la elaboración ni el contenido del mismo, que solo expresa la opinión de los autores.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
Nota al suplementoEste artículo forma parte del suplemento «Diagnóstico y tratamiento de las alteraciones del metabolismo de los triglicéridos: de la fisiopatología a la práctica clínica», que cuenta con el patrocinio de Akcea Therapeutics.