








Los triglicéridos (TG) son las moléculas más importantes para la reserva energética de nuestro organismo. Después de su síntesis hepática o intestinal a partir de los ácidos grasos son vehiculizados por los quilomicrones (QM) (origen intestinal) en el plasma o VLDL (origen hepático). Su catabolismo está determinado por la acción del complejo proteico de la lipoproteína lipasa (LPL) y de los receptores hepáticos encargados de su aclaramiento (RLDL y LRP-1). Las alteraciones en la producción o en el catabolismo se manifiestan como hipertrigliceridemia (HTG).
Las HTG se clasifican según su gravedad en leves-moderadas (150-885mg/dl), graves (>885mg/dl) o muy graves (>1.770mg/dl). Atendiendo su etiología pueden ser primarias y secundarias. Entre las principales formas primarias destaca el síndrome quilomicronémico familiar (SQF), forma muy severa debido a mutaciones del gen LPL o de proteínas asociadas. La mayoría de las HTG se deben a una coincidencia de factores de predisposición genética y ambientales.
Triglycerides (TG) are the most important molecules for the energy reserve of our body. After their hepatic or intestinal synthesis from fatty acids, they are carried by chylomicrons (QM (intestinal origin) or VLDL (hepatic origin) in plasma. Their catabolism is determined by the action of the lipoprotein lipase protein complex (LPL) and the hepatic receptors (RLDL and LRP-1) are responsible for their clearance are. Changes in the production or catabolism leads to hypertriglyceridaemia (HTG). The HTG are classified according to severity as, mild-moderate (150-885mg/dl), severe (>885mg/dl), or very severe (>1770mg/dl). They can be primary and secondary depending on origin. In the main primary form is highlighted Familial Chylomicronaemia Syndrome (CFS), a very severe form due to mutations in the LPL gene or associated proteins. Most HTG are due to a combination of genetic and environmental predisposing factors.
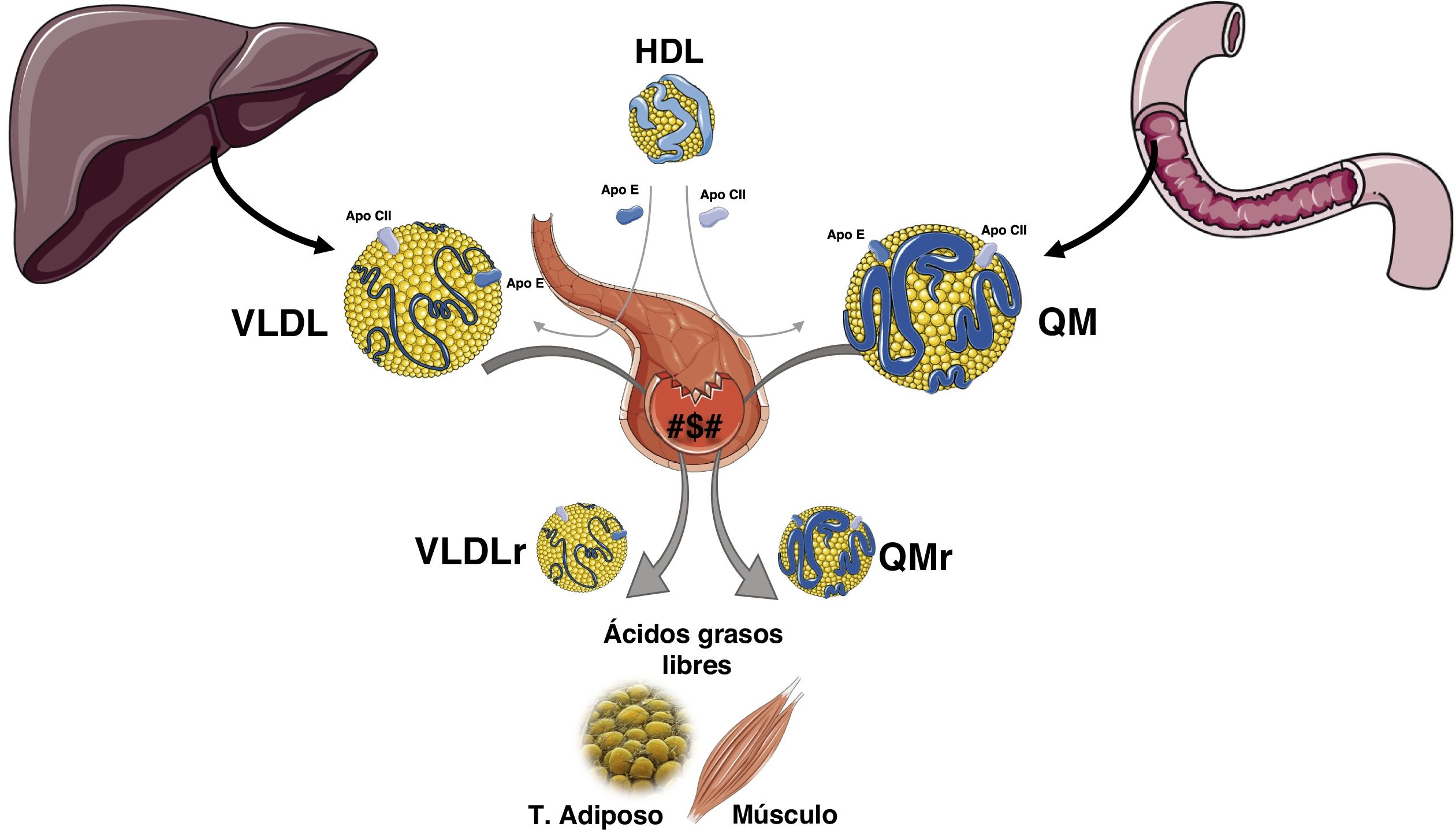
Los triglicéridos (TG) como todas las grasas son insolubles en el medio acuoso, por lo que deben ser transportados en el plasma como integrantes de las lipoproteínas, en las que son vehiculizados junto al colesterol, tanto libre como esterificado, a los fosfolípidos y a las apolipoproteínas. Además, son confinados en el núcleo de las partículas lipoproteicas debido a su carácter altamente apolar. Las lipoproteínas ricas en triglicéridos (LRT), es decir los quilomicrones (QM) y las lipoproteínas de muy baja densidad (VLDL), son complejos esféricos compuestos por lípidos altamente apolares (TG y ésteres de colesterol) que ocupan la zona central y, fosfolípidos (PL) y colesterol libre (CL) que dada su cierta polaridad se sitúan en la zona periférica junto a las apoproteínas de superficie1. Los TG exógenos que provienen de la dieta se incorporan en el intestino a los QM para su transporte, mientras que los TG de origen endógeno circulan en las VLDL derivadas del hígado2.
La formación de las VLDL en el hígado y de los QM en el enterocito es un proceso complejo que consiste en el ensamblaje de los distintos lípidos junto con apoproteínas específicas. La formación de las LRT comienza con la síntesis de TG, que en el intestino derivan de los ácidos grasos que provienen de la dieta, mientras que, en el hígado derivan de los ácidos grasos no esterificados que circulan por el plasma unidos a albúmina, de la lipogénesis de novo a partir de la glucosa y del catabolismo de las LRT endocitadas. Posteriormente, debido a una serie de acciones enzimáticas entre las que cabe destacar la de la diacilglicerol aciltransferasa se sintetizan los TG2,3. La proteína microsómica transferidora de triglicéridos (MTP) une los TG, el colesterol y los fosfolípidos, con isoformas específicas de tejido de la apolipoproteína B (apo B)4. La apo B-100 representa el producto completo del gen APO B y se sintetiza en el hígado, por lo que se secreta a la circulación en las VLDL, mientras que en los enterocitos se sintetizará una forma truncada por la creación de un codón stop una vez se ha sintetizado el 48% de la proteína, la apo B-48, que es la que constituirá los QM5. La síntesis de apo B es constitutiva, dado que se realiza de forma continua, siendo su regulación postraduccional. Una vez sintetizada, siendo la mayor proteína de nuestro organismo con un peso molecular de 512Kd, debe unirse a lípidos, dado que en caso contrario entra en un proceso de degradación a través de los proteasomas celulares.
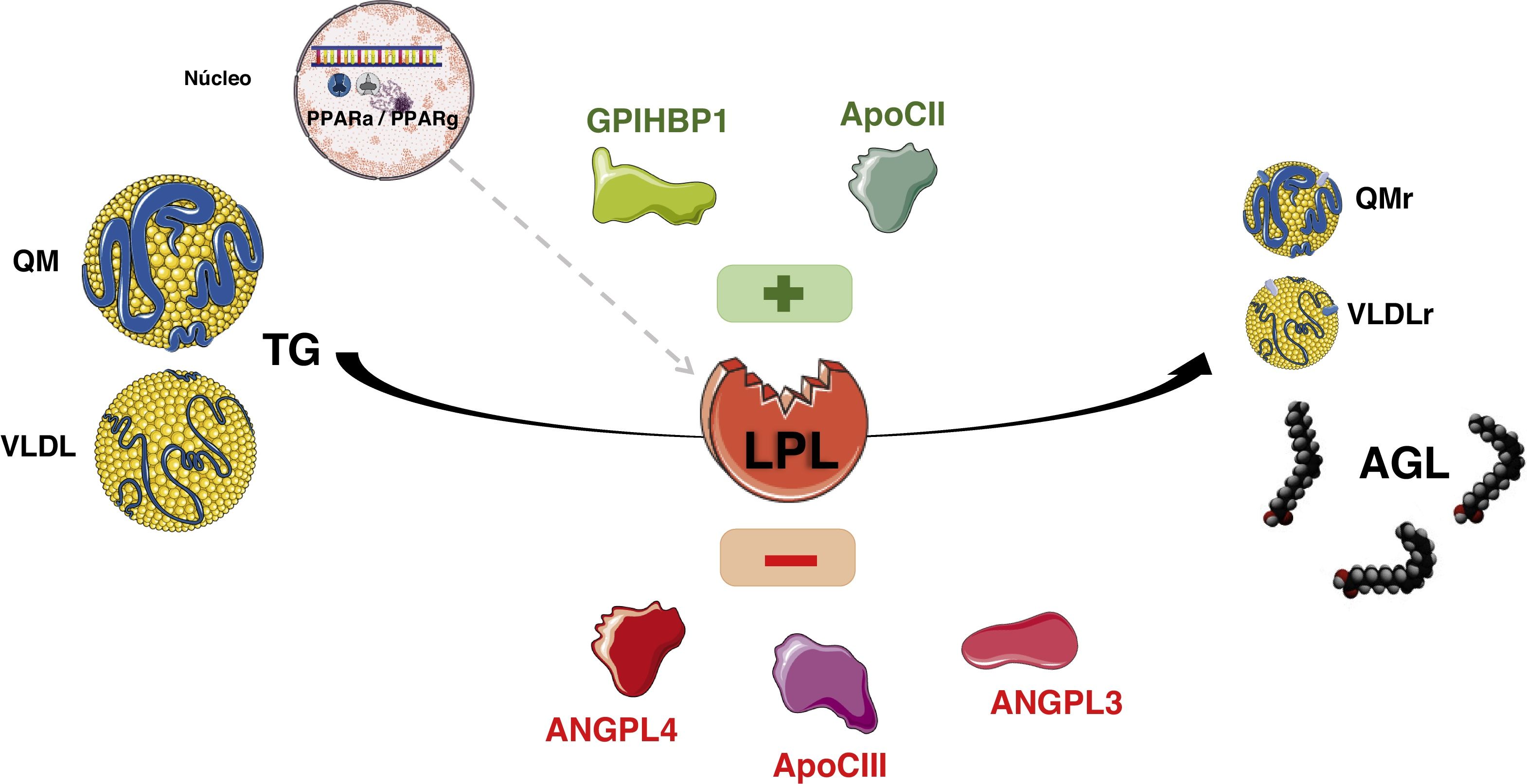
La síntesis de los QM se produce sobre todo en el período absortivo, y su concentración en el plasma alcanza su máximo a las 2-3h después de la ingesta (fase posprandial). De todas formas, la tasa de síntesis tanto de las VLDL como de los QM son muy variables, y a su vez están reguladas por la composición de la ingesta. Los QM pasan al plasma de forma indirecta a través de las vías linfáticas, mientras que las VLDL se secretan directamente a la circulación6. La hidrólisis de los QM maduros circulantes y de las VLDL está mediada por la actividad de la lipoproteína lipasa (LPL), que es una enzima fijada en la membrana de las células del endotelio capilar tanto del tejido adiposo como del muscular. La LPL, hidroliza los ácidos grasos de las LRT y permite que se internalicen en el tejido adiposo y muscular, para su almacenamiento y producción de energía, respectivamente. Esta acción de la LPL deriva en la producción de lipoproteínas residuales (remanentes) y de lipoproteínas de densidad intermedia (IDL). El aclaramiento de los remanentes de los QM vía hepática, requiere de la apo E, ya que la apo B-48 no tiene dominio de enlace en el receptor de lipoproteínas de baja densidad (RLDL). Los remanentes de los QM son aclarados a través del receptor denominado «proteínas relacionada con el receptor LDL» (LRP), mientras que los remanentes de VLDL e IDL se internalizan en los hepatocitos dado su contenido en apo E, que junto con apo B-100 actúan como ligandos para el RLDL. Aproximadamente un 50% de las IDL no son internalizadas y son remodeladas por la lipasa hepática (LH), generando así lipoproteínas de baja densidad (LDL). Están son internalizadas en las células periféricas, 80% a nivel hepático a través de los RLDL, cuya expresión celular está regulada, entre otros mecanismos, por la proproteína convertasa subtilisina kexina 9 (PCSK9) (fig. 1).

Existen varias proteínas que interactúan en la superficie endotelial controlando la actividad de la LPL. El factor de maduración de la lipasa 1 (LMF1) que es secretado por las células adiposas y miocitos, es una proteína acompañante que asegura que la LPL alcance su maduración y funcionalidad. La glycosylphosphatidylinositol anchored high density lipoprotein binding protein 1 (GPIHBP1) tiene la función de anclar la LPL al endotelio. La apo C2 activa la lipoproteína lipasa, mientras que la apo A5 es un cofactor estabilizador. La apo C3 inhibe la LPL, siendo un componente de las LRT. La angiopoietin-like 3 y 4 (ANGPTL3 y ANGPTL4) son proteínas recientemente descritas que también modulan la lipólisis afectando la concentración de las diversas familias lipoproteicas (fig. 2).
Metabolismo de las lipoproteínas ricas en triglicéridos ante trastornos metabólicos
A cualquier nivel de secreción de QM y VLDL la eficiencia, tanto de la lipólisis mediada por la LPL como la captación hepática de las partículas remanentes, determinará los niveles circulantes de TG en ayunas y posprandiales.
La concentración plasmática de TG es el resultado del equilibrio entre la tasa relativa de producción y de aclaramiento fraccional de las LRT, en las cuales se transportan los TG. A su vez, los TG puede hidrolizarse y eliminarse de las LRT, por lo que su concentración plasmática también depende del tamaño y de la composición de las LRT circulantes. La normolipidemia se mantiene cuando estos 2 procesos fisiológicos están en equilibrio, mientras que una alteración en este equilibrio conduce a hipertrigliceridemia (HTG)7. Ante una situación de incremento en la tasa de producción, puede haber un aumento compensatorio en la tasa de aclaramiento, pudiendo compensar la elevación de TG, regulando así su homeostasis. Sin embargo, en una situación de aumento de la entrada de TG endógenos o exógenos en el sistema y en la que el aclaramiento no puede compensarlo, puede desarrollarse HTG clínicamente significativa. Por tanto, la HTG puede surgir ante las siguientes situaciones: a) producción anormalmente alta sin un aumento compensatorio adecuado en el aclaramiento; b) aclaramiento gravemente deteriorado con una tasa de producción relativamente normal, y c) aumento de la producción y reducción del aclaramiento de forma simultánea2,8,9.
Muchos pacientes con HTG están afectos de síndrome metabólico, obesidad y/o diabetes mellitus tipo 2, que comparten la condición fisiopatológica de resistencia a la insulina. Esta condición clínica promueve el incremento en la secreción de VLDL, mediado por un exceso de ácidos grasos. En concreto, en presencia de resistencia a la insulina, se observa un incremento de los AG circulantes debido a un aumento de la lipólisis en el tejido adiposo secundario a una disminución en la señalización de la insulina. Al mismo tiempo aumenta la lipogénesis hepática ofreciéndose un incremento de sustrato para la síntesis de TG y finalmente de VLDL. A su vez esta situación, también puede conducir a un incremento de la secreción de QM, si bien el péptido similar al glucagón tipo 1 (GLP-1) inhibe la secreción de QM. Por otro lado, la apo C3 inhibe la eliminación de remanentes, por lo que cuando existe incremento en la secreción de VLDL y la apo C3 está elevada, la captación de partículas remanentes se ve reducida, agravando así la propia dislipidemia2,10.
En resumen, la resistencia a la insulina favorece la síntesis de LRT y reduce su aclaramiento plasmático, lo que resulta en HTG.
Hipertrigliceridemia y alteración global del metabolismo de las lipoproteínasEl metabolismo de las lipoproteínas tiene como objetivo fisiológico el transporte de substratos lipídicos como son los ácidos grasos. La formación de TG gobiernan dicho metabolismo, desde la estabilización de la apoproteína B tras su síntesis hasta el aclaramiento de las partículas finales del catabolismo lipoproteico, las LDL. Por ello, alteraciones en el metabolismo de los TG, incluso de tipo moderado, se asocian a modificaciones cualitativas del resto de los componentes de la cascada lipoproteica. Estudios con resonancia magnética nuclear (RMN) ponen de manifiesto, que incluso pequeñas alteraciones en las concentraciones de TG se acompañan de acúmulo de partículas remanentes de LDL más pequeñas y en mayor número, o de HDL de pequeño tamaño, distorsionadas y modificadas en su composición.
Por ello las HTG, incluso moderadas, deberían valorarse desde el punto de vista clínico más allá de lo que sus concentraciones plasmáticas sugieren11.
Clasificación de las hipertrigliceridemiasLa HTG en ayunas, se considera una condición clínica muy prevalente en la práctica clínica. Es muy probable que la HTG esté en aumento debido a la mayor prevalencia de obesidad y diabetes en nuestro medio. Clasificamos las HTG según su severidad y según su etiología.
Clasificación de las hipertrigliceridemias según la severidadSegún las concentraciones de TG plasmáticos alcanzadas dividimos las HTG en leves-moderadas, graves y muy graves (tabla 1)10.
Clasificación de las HTG según su gravedad
| HTG | TGmg/dl |
|---|---|
| Normal | <150 |
| Leve-moderada | 150-885 |
| Grave | >885 |
| Muy grave | >1.770 |
HTG: hipertrigliceridemia; TG: triglicéridos.
Fuente: adaptada adaptada de Laufs et al.10.
La HTG leve-moderada, bastante común, se considera con valores plasmáticos de TG entre 150-885mg/dl. Suele ser el resultado de factores ambientales secundarios como la dieta o el sobrepeso/obesidad y cierta predisposición genética, en general de tipo poligénico. En estas situaciones la HTG suele ser debida al acúmulo de VLDL y remanentes. Estas partículas suelen tener un tamaño inferior a los 70nm de diámetro pudiendo penetrar en la pared arterial y, por tanto, se consideran aterógenas. Es decir que las formas menos graves de HTG conllevan riesgo de aterosclerosis.
La HTG grave, que es menos común, se considera cuando los TG plasmáticos son superiores a 885mg/dl (10mmol/l). En general suelen coincidir factores desencadenantes externos con un condicionante genético de mayor trascendencia que en las formas menos graves, como mutaciones de genes principales del complejo enzimático LPL en forma heterocigota u oligogénica.
Existe una condición de HTG muy grave cuando los valores de TG son superiores 1.770mg/dl (20mmol/l). En estos casos suele encontrarse con mayor frecuencia una causa genética mayor como la homocigosidad para mutaciones en genes principales como la LPL y las proteínas asociadas.
Tanto en las formas graves como muy graves, a partir de unas concentraciones de TG de 1.000mg/dl, suelen acumularse QM. Estos, dado su tamaño tienen escasa capacidad aterógena y su principal riesgo es la producción de pancreatitis.
Esta clasificación es orientativa y las alteraciones generalmente asociadas a formas leves pueden manifestarse con excesos de TG de mayor gravedad debido a la aparición de factores exógenos desencadenantes.
Clasificación de las hipertrigliceridemias según su etiologíaHipertrigliceridemias primarias y secundariasDenominamos HTG primarias a las que son debidas a alteraciones propias del metabolismo lipídico y HTG secundarias a las producidas por factores exógenos desencadenantes.
Las formas primarias suelen ser el resultado de mutaciones monogénicas u oligogénicas siendo el factor genético el principal causante de la enfermedad.
La tabla 2 muestra la clasificación de las principales HTG primarias.
Hipertrigliceridemias primarias
| Hipertrigliceridemias graves (>885mg/dl, 10mmol/l) |
| Hipertrigliceridemias monogénicasDeficiencia de lipoproteína lipasa (mutación bialélica en el gen LPL)Deficiencia de Apo C2 (mutación bialélica en el gen APOC2)Deficiencia de Apo A5 (mutación bialélica en el gen APOA5)Deficiencia de LMF1 (mutación bialélica en el gen LMF1)Deficiencia de GPIHBP1 (mutación bialélica en el gen GPIHBP1) |
| Hipertrigliceridemias multigénicasSusceptibilidad genética compleja que incluyen:- Variantes genéticas heterocigotas raras de gran impacto para la hipertrigliceridemia monogénica (ver antes); y/o- Polimorfismos comunes acumulados de bajo impacto sobre la elevación de los TG, entre ellos, numerosos loci GWAS, incluidos APOA1-C3-A4-A5; TRIB1, LPL, MLXIPL, GCKR, FADS1-2-3, NCAN, APOB, PLTP y ANGPTL3- Hipertrigliceridemia infantil transitoria por deficiencia de GPD1 (mutación bialélica en el gen GPD1) |
| Hipertrigliceridemias leves-moderadas (177-875mg/dl, 2-9,9mmol/l) |
| Hipertrigliceridemia multifactorial o poligénicaSusceptibilidad genética compleja (ver antes)DisbetalipoproteinemiaSusceptibilidad genética compleja (ver antes), +Homocigosis para APOE E2/E2 oHeterocigosis de variante rara dominante de APOEHiperlipemia combinadaSusceptibilidad genética compleja (ver antes), +Acumulación de polimorfismos |
En las formas secundarias, si bien juegan un papel trascendental los factores ambientales, suele existir una interacción con condicionantes genéticos en general de tipo poligénico. En la tabla 3 mostramos las principales causas de HTG secundarias.
Hipertrigliceridemias secundarias (por orden alfabético)
| AcromegaliaAlcoholDerivación ilealDiabetes mellitus y resistencia a la insulinaDieta con alto balance de consumo de energía, alta en grasas y con alto índice glucémicoEmbarazo (especialmente en el tercer trimestre)Enfermedad renal (proteinuria, uremia o glomerulonefritis)Estrés, sepsis y quemadurasFármacos: antipsicóticos de segunda generación (clozapina y olanzapina), asparaginasa, beta-bloqueantes no cardioselectivos, ciclofosfamida, estrógenos por vía oral, glucocorticoides, isotretinoína, resinas de intercambio iónico, tamoxifeno y tiazidasGammapatías monoclonales y linfomasGlucogenosisHepatitis aguda (no fulminante)HipotiroidismoLipodistrofiasLupus eritematoso sistémicoObesidad y síndrome metabólicoSida y tratamiento antirretroviral con inhibidores de la proteasaSíndrome de Cushing |
Dicha interacción comportará diferencias en la expresión de la HTG. Habitualmente cursan de manera asintomática, y pueden acompañarse de C-HDL bajo, obesidad, diabetes mellitus y glucemia basal alterada12. En algunos casos la HTG es grave por la suma del componente poligénico y de factores ambientales, y puede llegar a comportarse y confundirse con el síndrome quilomicronémico familiar (SQF).
Síndrome de quilomicronemia familiarEs una forma muy severa de HTG primaria. El diagnóstico definitivo de este trastorno autosómico recesivo requiere del estudio genético de los siguientes 5 genes y de la detección de variantes raras, bialélicas (homocigotos o heterocigotos compuestos): LPL (que representa el 80% de casos), APOC2, APOA5, LMF1 y GPIHBP113 (tabla 2). De estas, las más comunes son las mutaciones de LPL y en segundo lugar las de GPIHBP112. En estos pacientes se objetiva un descenso drástico de la actividad LPL, lo que es un elemento importante para el diagnóstico.
El SQF cursa como complicación principal con pancreatitis aguda y dolor abdominal recurrente14. Se pueden observar también, xantomas eruptivos, lipidemia retinalis, esteatosis hepática y esplenomegalia.
La mayoría de los casos restantes de HTG severas o muy severas (más del 95% de los casos) se consideran de naturaleza multigénica15, por lo que también se denomina HTG multifactorial o poligénica, incluyendo la presencia de variantes heterocigotas raras en los 5 genes del SQF y/o variantes comunes acumuladas relacionadas con elevación de TG. Cabe destacar, que todas las causas secundarias (tabla 3) pueden agravar esta situación de HTG.
Hiperlipemias combinadasLa mayor parte de pacientes con HTG leve-moderadas suele presentar de forma simultánea elevaciones de colesterol, por lo que nos referimos a estas situaciones como hiperlipemias mixtas. Entre las formas primarias de hiperlipemias mixtas destacamos:
- –
La hiperlipemia familiar combinada, de causa poligénica, se manifiesta como HTG, pero también puede condicionar hipercolesterolemia por incremento de lipoproteínas de baja densidad (c-LDL). Se caracteriza por un fenotipo variable en un mismo paciente y en los miembros de la familia.
- –
La disbetalipoproteinemia tipo 3, se caracteriza por acúmulo de lipoproteínas de densidad intermedia (IDL) y remanentes de las LRT. En general se debe a una disfunción de la apo E que implica una menor afinidad por los receptores ApoB:E. Ello se debe a la homocigosis de la isoforma apo E2 (E2/E2) o la presencia de ciertas mutaciones raras dominantes del gen APOE.
El test de determinación de los TG se basa en la determinación de la concentración de glicerol en la muestra una vez hidrolizados los ácidos grasos. Existen circunstancias en las que se produce un incremento de la glicerolemia sin HTG que el test va a detectar como tal. Esta situación se denomina seudohipertrigliceridemia o hiperglicerolemia12. En esta situación hay unos TG séricos falsamente elevados por a un aumento de glicerol circulante; la principal causa de ello es la pérdida de función de la enzima glicerol cinasa.
Perspectivas futuras en la clasificación de las hipertrigliceridemiasMétodos como el anteriormente mencionado de la RMN, permite observar que las HTG pueden acompañarse de modificaciones en el resto de lipoproteínas relacionadas con un aumento del riesgo cardiovascular. En un futuro inmediato ciertas HTG deberán distinguirse de otras en base a su impacto sobre otras lipoproteínas como incremento en el número de partículas de LDL, menor tamaño de dichas partículas, presencia de HDL pequeñas o de HDL enriquecidas en TG.
FinanciaciónEste artículo ha sido financiado con una ayuda sin restricciones por Akcea Therapeutics. El patrocinador no ha intervenido en la elaboración ni en el contenido del mismo, que solo expresa la opinión de los autores.
Conflicto de interesesLM: honorarios como consejero o por conferencias de: Amgen, Sanofi, Mylan, Servier, Amryt, Amarin y Daiichi-Sankyo.
DI: honorarios por conferencias para Sanofi y Rubio.
Nota al suplementoEste artículo forma parte del suplemento «Diagnóstico y tratamiento de las alteraciones del metabolismo de los triglicéridos: de la fisiopatología a la práctica clínica», que cuenta con el patrocinio de Akcea Therapeutics.