




El síndrome de quilomicronemia familiar (SQF) es una entidad genética de herencia autosómica recesiva. Las mutaciones en genes (como APOC2, APOAV, LMF-1, GPIHBP-1) que codifican para proteínas que regulan la maduración, transporte o polimerización de lipoproteína lipasa-1 son las causas más comunes, pero no las únicas. El objetivo de este estudio fue reportar el primer caso documentado en el Ecuador.
Caso clínico: hombre de 38 años que presentó hepatoesplenomegalia crónica, trombocitopenia, atrofia pancreática e hipertrigliceridemia severa refractaria al tratamiento. Se realizó un análisis molecular por secuenciación de nueva generación que determinó una deficiencia de lipoproteína lipasa OMIM #238600 en homocigosis. La confirmación genética es necesaria a fin de poder establecer la etiología de HTGS para un adecuado manejo de esta patología.
Familial chylomicronemia syndrome (FCS) is a genetic entity with autosomal recessive inheritance. Mutations in genes (such as APOC2, APOAV, LMF-1, GPIHBP-1) that code for proteins that regulate the maturation, transport, or polymerization of lipoprotein lipase-1 are the most common causes, but not the only ones. The objective of this study was to report the first documented case in Ecuador.
Clinical caseA 38-year-old man presented with chronic hepatosplenomegaly, thrombocytopenia, pancreatic atrophy, and severe hypertriglyceridemia refractory to treatment. A molecular analysis was performed by next generation sequencing that determined a deficiency of Lipoprotein Lipase OMIM #238600 in homozygosis. Genetic confirmation is necessary in order to establish the etiology of HTGS for an adequate management of this pathology.
El síndrome de quilomicronemia familiar (SQF) es una enfermedad metabólica crónica que se caracteriza por una marcada elevación de triglicéridos y quilomicrones en sangre1. El SQF tiene origen genético, es de herencia autosómica recesiva y predispone a una elevada mortalidad por pancreatitis2,3.
El SQF se considera como una patología poco frecuente en el mundo y subestimada en el Ecuador2. La prevalencia global es de 1 por cada 1.000.000 de habitantes, y en centros especializados de alta complejidad, de hasta 1 por cada 300.000 habitantes3.
El SQF se identifica en la infancia debido a la hipertrigliceridemia severa (triglicéridos >10mmol/l) y pancreatitis aguda recurrente2,4. La clínica se distingue por la falta de respuesta a los tratamientos convencionales de reducción de triglicéridos más episodios repetidos de dolor abdominal, xantomatosis cutánea eruptiva, lipidemia retinalis, hepatoesplenomegalia y pancreatitis3. Además, presentan deterioro en el estado físico, emocional y cognitivo que impacta en la educación, en la situación laboral y en la calidad de vida5.
El diagnóstico del SQF se realiza con una prueba genética3. Las mutaciones patógenas bialélicas en los genes lipoproteína lipasa (LPL), apolipoproteína c2 (APOC2), glycosylphosphatidylinositol anchored high density lipoprotein binding protein 1 (GPIHBP1) o lipase maturation factor 1 (LMF-1) confirman la enfermedad. Se reportaron seis pacientes en los que se evaluaron autoanticuerpos contra las proteínas LPL o GPIHBP1, sin quilomicronemia familiar y con respuesta a inmunosupresores1.
El tratamiento está dirigido a reducir el triglicérido proveniente del intestino bajo la forma de quilomicrón. El Volanesorsen para los SQF6 es un oligonucleótido antisentido para disminuir la producción de la apoproteína C-III. La ApoC-III parece ser también un inhibidor de LPL-1 y especialmente un antagonista de apoE. Dentro de los efectos colaterales está la trombocitopenia7 y su uso está contraindicado en pacientes con una cifra de plaquetas menor de 140.000 (unidades). No se han reportado otras alternativas farmacoterapéuticas eficaces y seguras. La estrategia quirúrgica para disminuir una pancreatitis como la cirugía bariátrica8 y esplenectomía han sido alternativas extremas que incluso no se recomiendan por falta de evidencia9. El objetivo de este reporte fue describir la sintomatología y el diagnóstico confirmado de SQF que se presentó de forma atípica.
Caso clínicoUn hombre de 38 años con antecedentes médicos de diabetes mellitus tipo2 e hipotiroidismo; en la historia familiar se destacó la hermana, que murió a los 25años a causa de pancreatitis necrosante.
Se registraron triglicéridos elevados (560mg/dl) por primera vez a los 27años y tuvo varias hospitalizaciones por hipertrigliceridemia, con niveles que llegaron hasta 3.000mg/dl, sin presentar pancreatitis ni diagnóstico específico en los egresos hospitalarios. El paciente tenía un índice de masa corporal (IMC) de 23kg/m2 y signos vitales estables. No hubo hallazgos pertinentes durante el resto del examen físico.
Se realizó una ecografía abdominal que identificó el hígado heterogéneo de aspecto micronodular y esplenomegalia. Se definió como hepatopatía crónica.
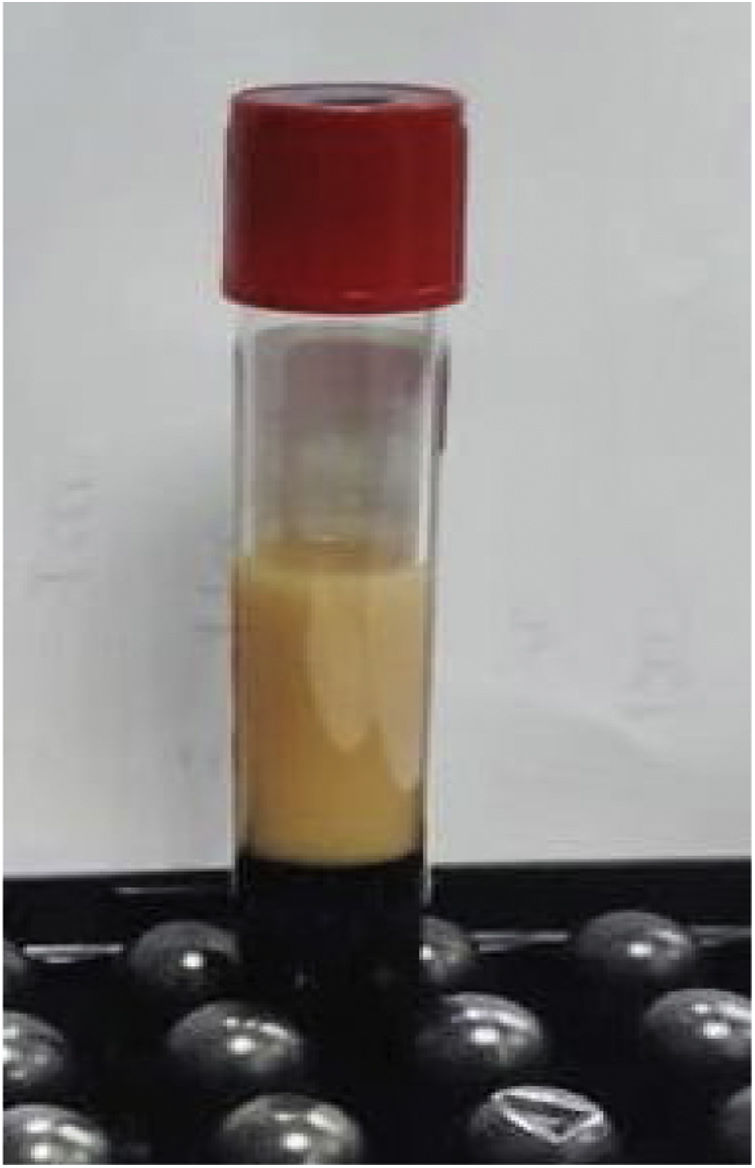
Los estudios de laboratorio iniciales revelaron niveles altos de triglicéridos, de 890mg/dl (fig. 1), colesterol total de 154mg/dl, colesterol LDL de 16mg/dl, colesterol HDL de 12mg/dl y 115 000 plaquetas por microlitro (μl).

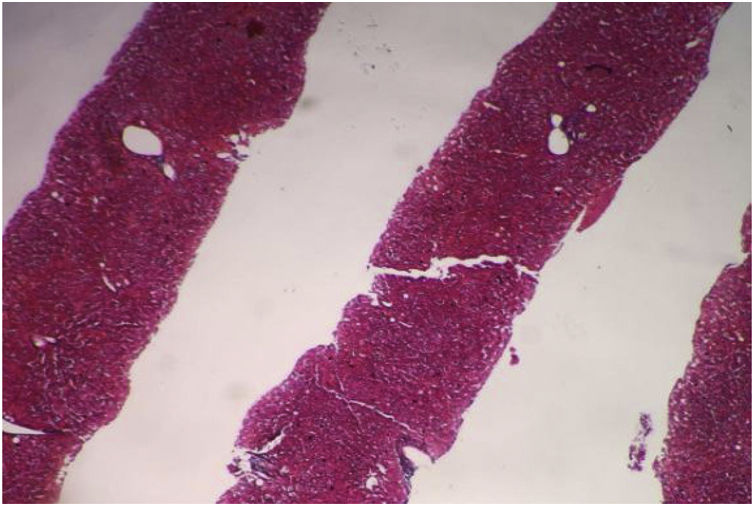
Se realizó una tomografía contrastada de abdomen que identificó el páncreas atrófico, el bazo aumentado de tamaño (hepatoesplenomegalia), sin líquido libre en la cavidad abdominal. Se decidió realizar biopsia hepática percutánea guiada por ecografía para la valoración de fibrosis debido a la discordancia entre los estudios de imagen (fig. 2).

Se realizó el estudio histopatológico, que identificó el parénquima hepático con estructura general conservada: se identificaron ocho espacios portales entre completos e incompletos. Los espacios portales y las estructuras ductales no presentaron alteraciones, y las estructuras vasculares no se observaron dilatadas. Los hepatocitos guardaron su habitual disposición radiada, las venas centrales no presentaron alteraciones y no se identificó fibrosis.
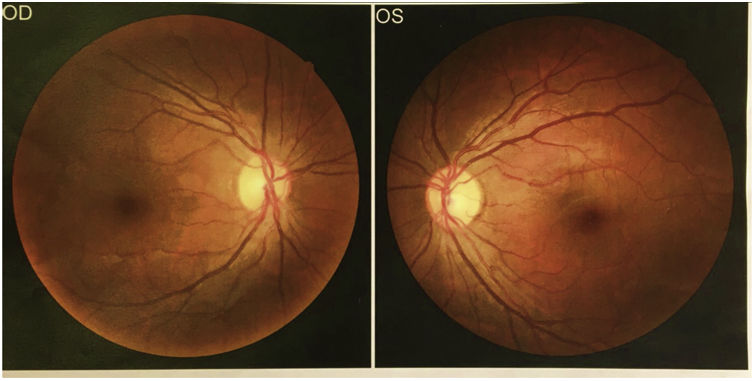
Se consultó a oftalmología para la realización de fondo de ojo, que no identificó lipidemia retinal (fig. 3).

Por la historia de múltiples ingrasos por hipertrigliceridemia y la respuesta refractaria al tratamiento hipolipemiante se realizó el diagnostico presuntivo de SQF, aunque el paciente negó consanguinidad entre los padres.
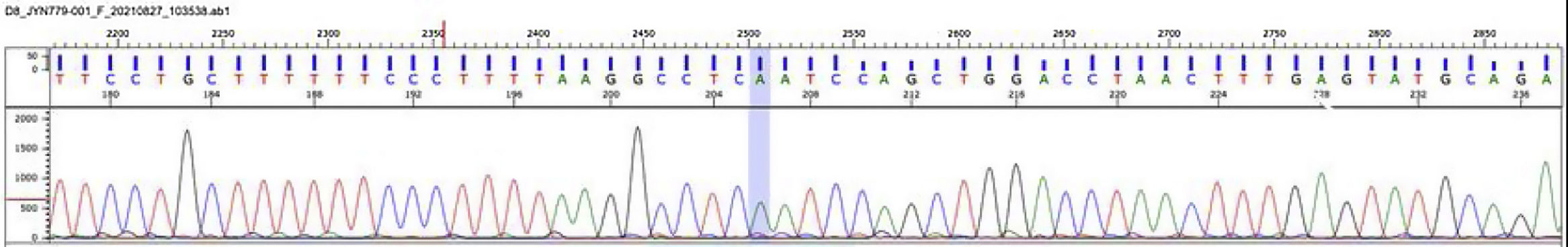
Se realizó el estudio molecular por secuenciación de nueva generación de los genes LPL, APOA5, APOC2, GPIHBP1 y LMF1. El análisis identificó una mutación homocigota en el gen LPL (lipoproteína lipasa, OMIM* 609708): c.547G>A (pAsp183Asn) [dbSNPrs118204064] (fig. 4). Esta mutación es considerada patogénica y se ha encontrado como responsable de deficiencia de lipoproteína lipasa10.
![Déficit de lipoproteína lipasa, electroferograma: estudio molecular por secuenciación de nueva generación de los genes LPL, APOA5, APOC2, GPIHBP1 y LMF-1. El análisis identificó una mutación homocigota en el gen LPL (lipoproteína lipasa, OMIM* 609708): c.547G>A (pAsp183Asn) [dbSNPrs118204064]. Fuente: Mendelics Analise Genomica S, Sao Paulo, Brasil.](https://static.elsevier.es/multimedia/02149168/0000003400000006/v2_202302030905/S0214916822001115/v2_202302030905/es/main.assets/gr4.jpeg?xkr=ue/ImdikoIMrsJoerZ+w997EogCnBdOOD93cPFbanNcxT+B4FjbiPdoCJto3d5S/Ch6vvG+Cp80/Jya7nfY0UqoFJQkREunQIvRoO1ZmxjZp0Bp4vBRLdiU9qkIRNn2hMnUySrJsBtAIxu5SHdfLpf0wDgq93YTyors27cZ6IOU6YZ6F7wEIqx7ItKnz/Ta7DvPgMV4/bZBN3Xmq3WHcK2lUS/8247ZpEmk7UaB11jaSyLzKH+7V0K6nqkF0SmXu3Dd/CFnfHlcK6WAOpB9SpN05oRhzP0ZHtgzBGTCwPqY= "Déficit de lipoproteína lipasa, electroferograma: estudio molecular por secuenciación de nueva generación de los genes LPL, APOA5, APOC2, GPIHBP1 y LMF-1. El análisis identificó una mutación homocigota en el gen LPL (lipoproteína lipasa, OMIM* 609708): c.547G>A (pAsp183Asn) [dbSNPrs118204064]. Fuente: Mendelics Analise Genomica S, Sao Paulo, Brasil.")
Déficit de lipoproteína lipasa, electroferograma: estudio molecular por secuenciación de nueva generación de los genes LPL, APOA5, APOC2, GPIHBP1 y LMF-1. El análisis identificó una mutación homocigota en el gen LPL (lipoproteína lipasa, OMIM* 609708): c.547G>A (pAsp183Asn) [dbSNPrs118204064].
Fuente: Mendelics Analise Genomica S, Sao Paulo, Brasil.
Se empezó tratamiento desde hace 4 años con metformina, inhibidores de la dipeptidil peptidasa 4 e insulina, gemfibrozilo, estatinas y ácido fenofíbrico. Además de la dieta baja en grasas y el control adecuado de comorbilidades. Los estudios de laboratorio actuales de triglicéridos fueron similares a los iniciales debido a la falta de una terapia eficaz y segura.
DiscusiónEn esta investigación se reportó el primer caso con SQF de Ecuador, que se integra en los pocos descritos a nivel mundial2,5. El paciente presentó hipertrigliceridemia y afectación esplénica sin pancreatitis, una sintomatología atípica. El análisis genético encontró una mutación homocigota en el gen LPL.
Se han descrito varios casos de SQF con otras mutaciones y caracterizados por la presencia de pancreatitis. Truninger et al.11 describieron dos hermanos en Suiza que tuvieron SQF debido a mutaciones heterocigotas compuestas en el gen LPL; ambos presentaron pancreatitis aguda recurrente. Ahmad y Wilson12 reportaron una paciente pediátrica en Estados Unidos con SQF que presentó pancreatitis aguda, tuvo mutaciones en el gen GPHBP1 y respondió bien a una dieta extremadamente baja en grasas y rica en triglicéridos de cadena mediana. Zhang et al.13 reportaron dos hermanos de China que presentaron signos de SQF en las primeras semanas de vida y tuvieron mutaciones en el gen LPL; los niveles de lípidos en plasma bajaron luego de intervenciones en sus dietas. Pinilla-Monsalve et al.14 reportaron una paciente colombiana de 50años con SQF que tuvo mutaciones en el gen APOC e historia médica de episodios severos y recurrentes de pancreatitis, sin respuesta al uso de agentes farmacológicos. Ueda et al.15 estudiaron tres hermanos de Estados Unidos con SQF que tuvieron mutación en el gen LPL e iniciaron con síntomas en la adolescencia, presentaron pancreatitis recurrente y respondieron a una dieta con restricción de lípidos. Cefalù et al.16 reportaron un paciente italiano con SQF con inicio de síntomas desde antes de los 10años, se identificaron mutaciones en el gen LPL y además tuvo pancreatitis recurrente. Este paciente presentó buena respuesta frente al tratamiento con lomitapida.
El SQF debe sospecharse con valores de triglicéridos mayores a 885mg/dl. Se propone usar el puntaje de Moulin basado en características clínicas, que con valores mayores de 10 indica una alta probabilidad de esta patología3. El SQF debe diferenciarse del síndrome de quilomicronemia multifactorial (SQM), que se presenta también con hipertrigliceridemia severa y pancreatitis pero difiere en la edad de presentación. El primero aparece usualmente en la infancia o en la adolescencia, mientras que el segundo lo hace más frecuentemente en la adultez. El SQM también puede estar acompañado de otras patologías metabólicas, como diabetes mellitus y esteatosis hepática17. La heterogeneidad de aparición de pancreatitis y la falta de conocimiento de la enfermedad contribuyen a una atención inadecuada2,18.
La evaluación del paciente reportado fue atípica, y el seguimiento a largo plazo de futuras complicaciones aún queda por investigar. El manejo multidisciplinario y la detección oportuna con el análisis genético frente a una dislipidemia refractaria al tratamiento convencional permitieron el diagnóstico del primer caso de SQF reportado en el Ecuador. El abordaje interdisciplinario y el diagnóstico molecular mediante la identificación de mutaciones en genes candidatos permitirían un manejo adecuado con las opciones terapéuticas disponibles.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.