




El objetivo de esta publicacion es profundizar y dar a conocer a la comunidad médica la existencia del síndrome de QT corto (SQTC), descrito recientemente, como una enfermedad eléctrica primaria del corazón, asociada a un riesgo elevado de muerte súbita por arritmias ventriculares malignas y a episodios de fibrilación auricular. Se enfatiza que este cuadro se presenta de forma general en jóvenes sin enfermedad estructural cardíaca. Las mutaciones identificadas en los genes codificadores de la síntesis de los canales de potasio implican una ganacia de su función y, por consiguiente, un acortamiento en la duración del potencial de acción en el período refractario auricular y ventricular. El electrocardiograma muestra un intervalo QT menor de 300 ms con ondas T puntiagudas, aunque con carácter intermitente. El diagnóstico precisa de la exclusión de las causas fisiológicas y extrínsecas que acortan el intervalo QT. Se concluye que el SQTC, aunque infrecuente, debe ser del dominio del personal médico especializado por las implicaciones pronósticas que reviste y que la implantación de un desfibrilador automático es el tratamiento de primera línea en los pacientes sintomáticos. Los fármacos antiarrítmicos del grupo III, en especial la quinidina, representan otra opción terapéutica.
The target of this publication is to make a dipper view and to let know to the scientific medical sommunity about the recently description of the short QT syndrome as a heart primary electrical disease, asociated to a high sudden death risk due to malignat ventricular arrhythmias and to atrial fibrillation episodes. It is emphasized that this situation can be frequently found in young people, without a structural cardiac disease. The identified mutations in synthesis codificator genes of potassium channels implies a profit of its function, and therefore a reduction in the potencial action duration and in atrial and ventricular refractory periods. The electrocardiogram shows a QT interval smaller than 300 ms with sharp T wave, although with an intermittent character. The diagnosis requires the exclusion of physiological and extrinsic causes that make shorter the QT interval. It is concluded that the short QT syndrome, though unfrequent, must be widely known by the specialized medical staff because of the prognosis implications that it has and because the implantion of an automatic desfibrillator constitutes first line therapy in symptomatic patients. The a class III antiarrhythmic drugs, specially quinidine, represent another therapeutic option.
El síndrome de QT corto (SQTC) es una enfermedad eléctrica primaria del corazón de base genética, caracterizada, eléctricamente, por una dismi nución en la duración del intervalo QT (IQT), y, clínicamente, por la presencia de arritmias cardíacas y elevada asociación a muerte súbita.
Este síndrome descrito recientemente se inicia a edades relativamente tempranas de la vida y se asocia a episodios de fibrilación auricular y a un riesgo elevado de muerte súbita, y muchas veces el primer síntoma es el episodio letal. Su diagnóstico no precisa de medios sofisticados, son suficientes el electrocardiograma y la historia clínica, herramientas sencillas y accesibles. Por tratarse de un fenómeno funcional, y debido a su frecuente carácter intermitente, no siempre se observa en los trazados electrocardiográficos habituales y se debe buscar en estudios Holter o en secuencias eléctricas.
Breve reseña históricaLo describieron por vez primera en el año 2000 Gussak et al1, los cuales identificaron un IQT corto en 3 miembros de una misma familia, de los cuales uno había presentado varios episodios de fibrilación auricular. Pero la relación definitiva entre el SQTC y la muerte súbita la plantearon Gaita et al2 en el año 2003.
En el año siguiente, Brugada et al3 describen en individuos afectados de una misma familia la primera mutación causante del SQTC, ubicada en el gen codificador del componente rápido de la corriente tardía rectificadora de potasio (IKr)3. Poco tiempo después, Bellocq et al4 informan en un único paciente, con IQT corto y muerte súbita abortada, una mutación en el gen que codifica el componente lento de la corriente rectificadora tardía de potasio (IKs).
La tercera mutación en el SQTC la identificaron Priori et al5 en el 2005, localizada en el gen KCNJ2, causante de las corrientes de repolarización de potasio IK1.
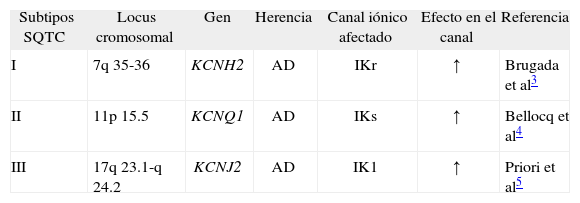
Bases genéticas y electrofisiológicasHasta la fecha, se han ligado 3 genes al SQTC: KCNH2, KCNQ1, KCNJ2, todos ellos codificadores de la síntesis de canales iónicos de K+ (tabla 1).
Mutaciones en el SQTC
| Subtipos SQTC | Locus cromosomal | Gen | Herencia | Canal iónico afectado | Efecto en el canal | Referencia |
| I | 7q 35-36 | KCNH2 | AD | IKr | ↑ | Brugada et al3 |
| II | 11p 15.5 | KCNQ1 | AD | IKs | ↑ | Bellocq et al4 |
| III | 17q 23.1-q 24.2 | KCNJ2 | AD | IK1 | ↑ | Priori et al5 |
AD: autosómica dominante; SQTC: síndrome de QT corto.
Este gen expresa una proteína del canal IKr, encargada de la corriente de salida rápida de K en el inicio de la fase 3 del potencial de acción. Se han identificado 2 mutaciones en familias no relacionadas. En ambas hay una sustitución de aspargina por lisina en el codón 588 del poro del canal.
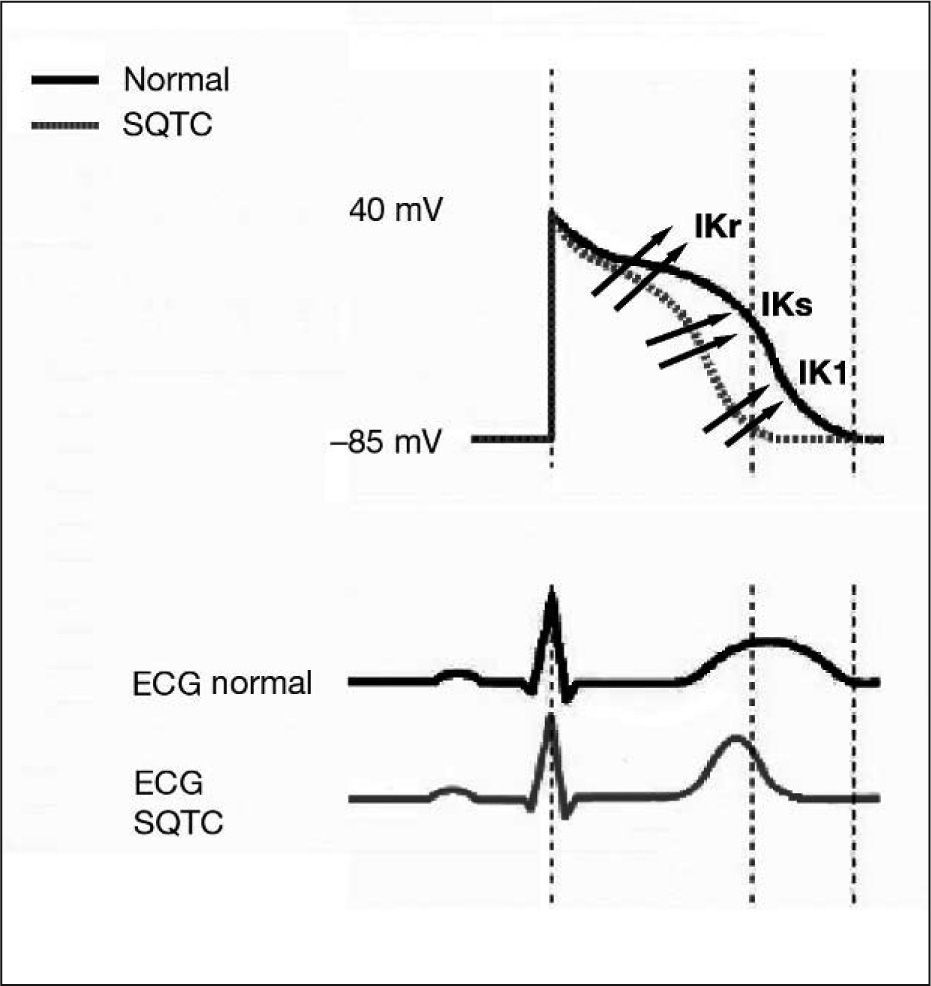
La mutación suprime la inactivación del canal, con el incremento consiguiente de la corriente neta de salida de K+. La ganancia de función del canal IKr genera un acortamiento en la duración del potencial de acción y en el período refractario, tanto auricular como ventricular, lo que justifica la alta asociación de fibrilación auricular y de arritmias ventriculares en este síndrome3 (fig. 1).
KCNQ1 en el síndrome de QT corto (SQTC). Ganancia de función en las corrientes de salida IKr, IKr e IK1, con acortamiento en la duración del potencial de acción en el SQTC.")
El gen KCNQ1 codifica una proteína del canal IKs. La primera mutación en este gen la identificaron Bellocq et al4 en un varón de 70 años con fibrilación ventricular e IQT corto (290ms) posreanimación. De forma más reciente, el grupo de Brugada et al identificó una mutación de novo en una recién nacida de 38 semanas con fibrilación auricular con respuesta ventricular lenta e IQT corto6. Ambas mutaciones implican una ganancia de función del canal IKs y, por tanto, un acortamiento de la refracteriedad auricular y ventricular.
KCNJ2El tercer subtipo de SQTC es producido por una mutación en el gen KCNJ2, el cual codifica una proteína del canal encargada de la corriente IK1. La mutación se indentificó en una niña de 5 años y en su padre. Ambos mostraban un IQT no excesivamente corto y una onda T con una fase terminal anormalmente rápida; su padre refería además cuadros presincopales y palpitaciones desde la edad de 15 años. Los efectos electrofisiológicos del canal mutante se traducen en una ganacia de función de éste, con la consiguiente aceleración de la repolarización tardía y el acortamiento significativo en la duración del potencial de acción5.
El SQTC es una enfermedad genética heterogénea, con un patrón de herencia autosómica dominante7. A pesar de un minucioso cribado genético, hay pacientes sintomáticos con IQT corto en los que no se ha identificado la mutación responsable. Al igual que en el síndrome de QT largo, la presentación clínica, los desencadenantes del episodio arrítmico y las opciones terapéuticas pudieran estar determinadas por la mutación genética8. Se necesitará de estudios futuros para identificar nuevos sitios mutantes, así como para establecer una correlación genotipo-fenotipo en el SQTC.
Todas las mutaciones implican una ganacia de función en una corriente de salida de K+ y, por tanto, un acortamiento en la duración del potencial de acción y en el período refractario auricular y ventricular. Los diferentes momentos de activación de las corrientes afectadas en cada subtipo de SQTC justifica las diferencias en la magnitud del acortamiento del IQT y en la morfología de la onda T.
Como la distribución de las corrientes iónicas en el miocardio no es homogénea, se especula que el acortamiento de la duración del potencial de acción en el SQTC tampoco lo es3,9. Éste se produce de forma preferencial en las células endocárdicas y epicárdicas, las que previamente tienen una duración menor del potencial de acción, lo que acentúa la dispersión de la repolarización entre éstas y las células M. Por otro lado, no se produce acortamiento en el período refractario de las células de Purkinje. La heterogeneidad resultante de la repolarización interventricular e intraventricular generará un sustrato idóneo para el desarrollo de arritmias ventriculares malignas10–12.
Se desconocen las influencias del sistema nervioso autonómico en estas mutaciones. El ejercicio, el reposo y el estrés desencadenan el episodio arrítmico13,14. En un joven de 16 años con el diagnóstico de SQTC y desfibrilador automático implantable, se produjo una fibrilación ventricular durante el sueño, desencadenado por un complejo ventricular prematuro con intervalo de acoplamiento corto; en cambio, su padre, con igual diagnóstico, murió de forma súbita de una fibrilación ventricular durante un esfuerzo físico intenso15. Ello indica que el disparador del episodio arrítmico puede estar determinado por el tipo de mutación.
Recientemente, Antzelevitch et al16 describieron una nueva mutación en 3 individuos con fenotipo Brugada, IQT corto (< 360ms) e historia personal y familiar de muerte súbita. La mutación se ubicó en los genes encargados de la síntesis de las subunidades α y β del canal lento de Ca+2. La mutación resulta en una pérdida de función del canal, con la disminución consiguiente en la entrada del ion en la fase 2 del potencial de acción y el acortamiento en los períodos refractarios. La adaptación del IQT a la frecuencia cardíaca es patológica, al igual que en los restantes subtipos de SQTC. La quinidina normalizó el IQT y previó la reinducción de arritmias ventriculares en el estudio electrofisiológico. Sin embargo, a todos los pacientes se les implantó desfibrilador automático implantable debido al riesgo elevado de muerte súbita16.
Manifestaciones clínicasLa presentación clínica es altamente variable, desde formas asintomáticas hasta fibrilación auricular paroxística o permanente, síncope, arritmias ventriculares y muerte súbita1,2,13. La edad de inicio puede ser muy temprana, tanto como en el primer año de vida, y ser la causa de algunos casos de muerte súbita en el lactante2. La entidad muestra un porcentaje elevado de una historia familiar de muerte súbita y/o fibrilación auricular.
La inducibilidad de las arritmias ventriculares y de la fibrilación auricular en el estudio electrofisiológico es alta. La no inducibilidad de las arritmias ventriculares no excluye el riesgo futuro de experimentar el episodio. Por esta razón no es el único factor de estratificación de riesgo en estos pacientes13,15.
Alteraciones electrocardiográficas- 1.
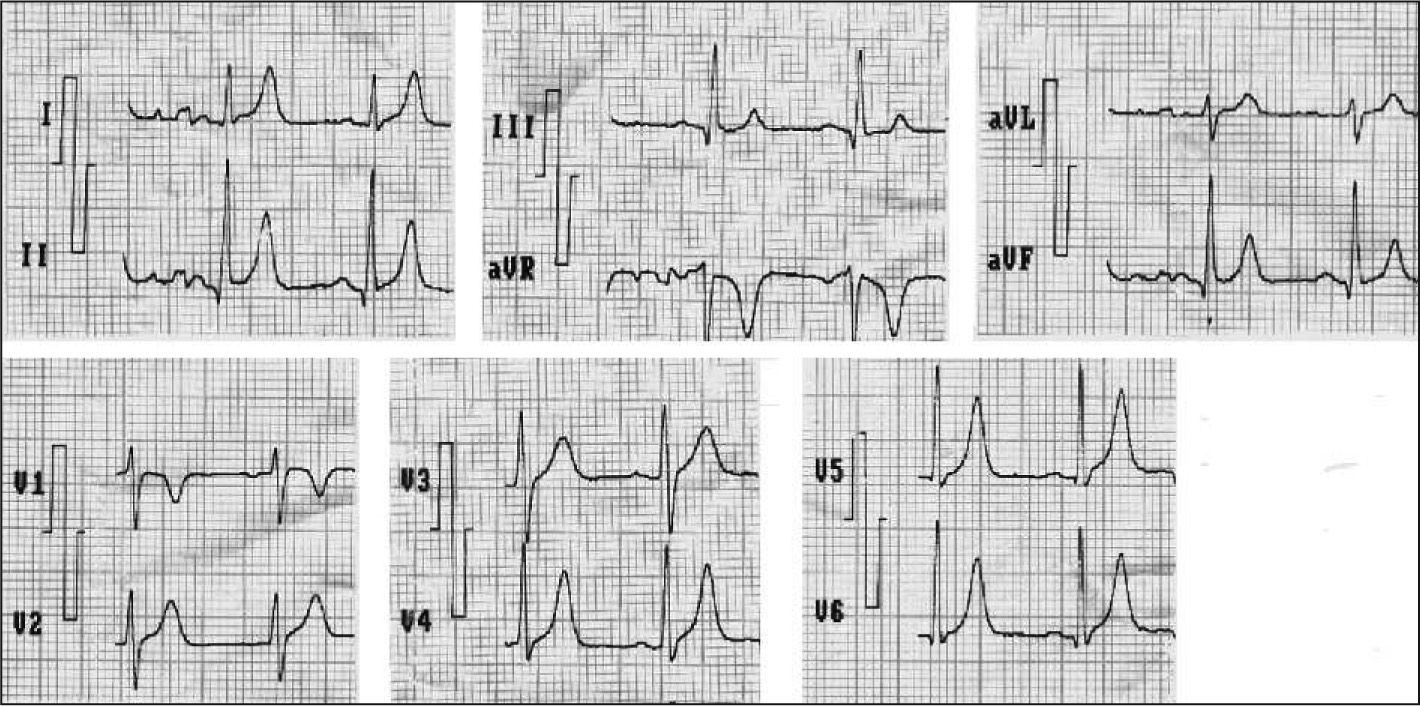
IQTc < 300ms. Con un rango de 220–300ms para una frecuencia cardíaca entre 60 y 85 latidos por minuto (lpm). Este acortamiento puede ser intermitente (fig. 2). El SQTC tipo 3 no muestra un acortamiento tan acentuado, mide alrededor de 360ms.
El acortamiento inadecuado o la prolongación del IQT secundario a las variaciones de la frecuencia cardíaca obliga a medir y a corregir este intervalo a valores de frecuencia entre 60 y 85 lpm5,13.
- 2.
Pérdida de la adaptación del IQT a las variaciones de la frecuencia cardíaca, con una disminución de la pendiente de la curva IQT/frecuencia cardíaca, que puede ser un peldaño fundamental en el diagnóstico del SQTC2,17.
- 3.
El 50% de los pacientes exhiben ondas T puntiagudas y simétricas en derivaciones precordiales, que se inician inmediatamente después de la onda S, con segmento ST muy corto o prácticamente ausente. En el SQTC tipo 3, las ondas T son asimétricas, con una rama ascendente normal y una fase terminal excesivamente rápida de la rama descendente. Esta apariencia peculiar de la onda T puede estar en relación con la aceleración súbita de la fase final de la repolarización del potencial de acción, mediado por las corrientes IK15,13,18.
- 4.
Intervalo largo del final de la T al inicio de la P y ausencia de la onda U10.

Hasta la fecha se ha diagnosticado aproximadamente a 40 pacientes con SQTC7. Debido a que la longitud del IQT es un parámetro clínico modificable por los cambios de la frecuencia cardíaca, el acortamiento fisiológico de éste debe diferenciarse de los acortamientos de causa extrínseca y del SQTC en sí. Dentro de las causas extrínsecas que acortan el IQT se citan las siguientes: taquicardia, hiperpotasemia, hipercalcemia, acidosis, intoxicación digitálica, hipertermia y valores elevados de catecolaminas, acetilcolina y testosterona13,19–21.
Para establecer el diagnóstico de SQTC se precisa:
- 1.
IQT corto en ausencia de causa extrínseca.
- 2.
Fibrilación auricular y/o fibrilación ventricular, documentadas o con síntomas en relación con estas arritmias.
- 3.
Antecedentes familiares de muerte súbita y/o SQTC.
La existencia de un IQT corto aislado, en ausencia de síntomas o historia familiar, debe considerarse un signo electrocardiográfico de QT corto y no el síndrome en sí, tal como sucede con el síndrome de Brugada y el electrocardiograma tipo Brugada. No debe cometerse el error de diagnosticar una entidad fuera del contexto clínico. El desfibrilador automático implantable en una paciente con SQTC correctamente diagnosticado salva una vida, mientras que en un paciente con signo de QT corto representa una carga humana, económica y social.
A todos los pacientes se debe realizar: prueba de esfuerzo; Holter (seguimiento electrocardiográfico) de 24 o 48h; estudio electrofisiológico para determinar el período refractario auricular y ventricular, y para un protocolo de inducción de taquiarritmia ventricular; así como el cribado genético. Este último aspecto no está extendido a todos los servicios13,21.
TratamientoEl riesgo de muerte súbita en las escasas familias estudiadas es alto, por lo que se ha implantado desfibrilador automático en la mayoría de los pacientes sintomáticos2,10,13,22. Un problema frecuente es el tratamiento inapropiado de desfibrilación, secundario al sobresensaje de la onda T. Esta situación se resuelve mediante la reprogramación con algoritmo de supresión de la onda T, y con la adición de quinidina, la cual normaliza el IQT y disminuye la amplitud de la onda T13,17,22.
El tratamiento farmacológico para prevenir la muerte súbita debe tratarse con precaución. Sólo se ha utilizado en individuos con mutaciones en los canales IKr17. Se considera tratamiento de segunda línea en pacientes con riesgo alto de muerte súbita, de existir negativa para el implante del dispositivo o imposibilidad para realizarlo. Sin embargo, el tratamiento farmacológico es de utilidad en la supresión de los episodios de fibrilación auricular y el sobresensaje de la onda T, situaciones frecuentes en el SQTC.
De los fármacos con potencial efectividad en el SQTC, el sotalol y el ibutilide no prolongan el IQT en las investigaciones realizadas3,23,24. La flecainida, aunque lo prolonga, no disminuye la inducibilidad de fibrilación ventricular en la estimulación programada25. Sólo la quinidina resultó efectiva en prolongar el IQT, restituir la mala adaptación a los cambios de frecuencia cardíaca, disminuir la amplitud de la onda T y prevenir la inducción de fibrilación ventricular en el estudio electrofisiológico17,22,24,25. La propafenona se usó con éxito en 2 pacientes para suprimir los paroxismos de fibrilación auricular25.
Se concluye que el SQTC, aunque infrecuente, debe tenerse en consideración como posibilidad diagnóstica ante toda persona joven con episodios de muerte súbita y/o episodios de fibrilación auricular con elementos electrocardiográficos característicos o indicativos. Se deben descartar las causas extrínsecas de acortamiento del IQT. El cardiodesfibrilador automático implantable es el tratamiento de primera línea en pacientes con arritmias ventriculares malignas. Los fármacos antiarrítmicos del grupo III, en especial la quinidina, representan una opción terapéutica válida.