




Entre el 30 y el 50% de los sujetos con diagnóstico genético de hipercolesterolemia familiar (HF) heterocigota presentan xantomas tendinosos (XT), pero el mecanismo por el cual unos sujetos HF desarrollan XT y otros no se desconoce. Previamente, nuestro grupo de investigación ha demostrado que los macrófagos de sujetos HF con y sin XT desarrollan una respuesta inflamatoria diferente frente a lipoproteínas de baja densidad oxidadas (LDLox). Por ello, el objetivo de este trabajo fue analizar la expresión génica de diversas moléculas inflamatorias que podrían estar involucradas en la aparición y desarrollo de XT.
Material y métodosSe seleccionó a 10 pacientes con diagnóstico genético de HF, en los que se midió el diámetro anteroposterior del tendón de Aquiles mediante ecografía de alta resolución. Se aislaron sus monocitos a partir de 40 ml de sangre periférica. Una vez diferenciados a macrófagos, se suplementaron con 50 μg/ml de LDLox durante 1, 3, 6 y 18 h. Mediante RT-PCR en tiempo real, se analizó la expresión de los genes PPARγ, IL-8, IL-1β, CXCL3, triptasa, NF-κBIA y TNF-α.
Resultados y conclusiónLos sujetos HF con XT (HF XT+) mostraron una tendencia a sobreexpresar el gen IL-8 tras 18 h de incubación con LDLox, mientras que el grupo de sujetos HF sin XT (HF XT–) tendió a sobreexpresar el gen TNF-α tras 1 h de incubación con LDLox. El gen CXCL3 se sobreexpresó significativamente en todos los tiempos de incubación en el grupo HF XT+. Además, se halló una correlación positiva entre la expresión de CXCL3 y el tamaño del tendón de Aquiles, que fue máxima a 3 h del tratamiento con LDLox (R=0,782; p=0,008). Estos resultados sugieren que CXCL3 podría desempeñar un papel importante en la etiología de los xantomas, y se puede considerar como un posible marcador predictor de estos depósitos lipídicos.
Approximately 30%-50% of patients with genetic diagnosis of heterozygous familial hypercholesterolemia (FH) present tendon xanthomas (TX), but the mechanism by which some subjects develop TX and others do not is unknown. Previously, we have shown that macrophages of FH subjects with and without TX develop a different inflammatory response to oxidized LDL (oxLDL). Therefore, the objective of this work was to analyze the gene expression of several inflammatory molecules that could be involved in the onset and development of TX.
Material and methodsTen FH patients were selected, and the antero-posterior Achilles tendon diameter was measured with high resolution sonography. Their monocytes were isolated from 40 ml of peripheral blood. When they were differentiated to macrophages, were supplemented with 50 μg/ml of oxLDL for 1, 3, 6 and 18 hours. The gene expression of PPARγ, IL-8, IL-1β, CXCL3, tryptase, NF-κBIA and TNF-α was analyzed with real time RT-PCR.
Results and conclusionThe FH subjects with TX (FH TX+) showed a tendency to over-express IL-8 gene after 18 h of incubation with oxLDL, while FH subjects without TX (FH TX–) tended to overexpress TNF-α gene after 1 h of incubation. CXCL3 gene was significantly over-expressed at all incubation times with oxLDL in FH TX+ group. Furthermore, a positive correlation was found between CXCL3 gene expression and Achilles tendon size, being maximum at 3h of treatment with oxLDL (R=0.782; p=0.008). These results would suggest that CXCL3 could play an important role in the ethiology of xanthomas and could be considered as a possible predictor marker of these lipid deposits.
La hipercolesterolemia familiar (HF) es una enfermedad monogénica causada por mutaciones en el gen del receptor de lipoproteínas de baja densidad (LDLR)1. Se caracteriza por elevadas concentraciones en plasma del colesterol unido a lipoproteínas de baja densidad (cLDL), depósitos de colesterol en tejido extravascular, como xantomas tendinosos (XT) y arco corneal, y mayor riesgo de enfermedad cardiovascular prematura2. En pacientes HF heterocigotos, la expresión clínica de la enfermedad es muy variable en términos de severidad, presencia de XT y edad de aparición de enfermedad cardiovascular, incluso compartiendo la misma mutación en el gen del LDLR3,4.
Los XT son depósitos de lípidos en los tendones que producen engrosamientos y tumoraciones. Están formados por células espumosas cargadas de lípidos, debido a la acumulación de LDL oxidada (LDLox) y tejido conectivo5,6. Son altamente específicos de HF en sujetos con altas concentraciones de cLDL, por lo que es recomendable incluirlos como un importante criterio diagnóstico clínico7. Aproximadamente del 30 al 50% de sujetos con diagnóstico genético de HF heterocigota presentan XT3,4,8. El mecanismo por el cual algunos sujetos HF desarrollan XT y otros no, incluso con concentraciones de cLDL similares y compartiendo la misma mutación en el gen LDLR, se desconoce3, pero suponen un mayor riesgo cardiovascular9. Aunque trabajos anteriores han sugerido que la presencia de XT podría estar controlada por un gen diferente del LDLR, éste aún no ha sido identificado10.
Nuestro grupo ha demostrado previamente que los macrófagos de sujetos HF con y sin XT desarrollan una respuesta inflamatoria diferencial frente a LDLox in vitro11. Por ello, la hipótesis de este trabajo fue que la sobreexpresión de ciertas quimiocinas y citocinas proinflamatorias frente a LDLox podría estar involucrada en la aparición y desarrollo de XT. Para evaluar nuestra hipótesis, se analizó el perfil de expresión de los genes PPARγ, NF-κBIA, IL-8, IL-1β, CXCL3, triptasa (TPS) y TNF-α en macrófagos de pacientes HF con y sin XT estimulados con LDLox durante diferentes tiempos.
Material y métodosSujetosSe seleccionaron 10 pacientes (6 varones y 4 mujeres) con diagnóstico genético de heterocigosidad para HF causada por mutaciones funcionales en el gen del LDLR y, por tanto, diagnóstico seguro de HF de acuerdo con los criterios establecidos en el programa internacional MEDPED. De ellos, 5 (3 varones y 2 mujeres) presentaron XT detectados antes de los 50 años, constituyendo el grupo HF XT+, y 5 (3 varones y 2 mujeres) carecían de XT, constituyendo el grupo HF XT-. El diagnóstico genético se llevó a cabo mediante la identificación de la mutación en el gen LDLR con la plataforma Lipochip®12. Ninguno de los pacientes estaba en tratamiento hipolipemiante en el momento de ser reclutado para el estudio. Todos los sujetos participantes firmaron un consentimiento informado y el estudio fue aprobado por el Comité de Ética del Instituto Aragonés de Ciencias de la Salud (I+CS), de acuerdo con la declaración de Helsinki de 1975, y con la revisión de octubre de 2000.
Perfil lipídicoSe obtuvo una muestra de sangre de cada paciente tras 12h de ayuno para realizar las medidas de colesterol total en plasma, colesterol unido a lipoproteínas de alta densidad (cHDL) y triglicéridos. Las medidas se realizaron con kits de diagnóstico comerciales (Boehringer-Mannheim, Indianapolis, IN, EE.UU.). El cLDL en plasma se calculó mediante la fórmula de Friedewald.
La lipoproteína(a) se determinó mediante inmunonefelometría con anticuerpos monoclonales (Beckman Coulter Inc., Miami, FL, EE.UU.).
Determinación del diámetro anteroposterior del tendón de AquilesSe determinó el diámetro anteroposterior del tendón de Aquiles (diámetro medio del tendón de Aquiles [DMTA]) mediante ecografía de alta resolución con un ecógrafo Acousting Imaging 5200S (Dornier, Phoenix, EE.UU.) y con una sonda LA 7,5/38 de 7,5MHz, con el procedimiento descrito previamente por nuestro grupo12. Para este estudio se consideró la presencia de xantoma cuando el DMTA fue mayor que la media ± 10 desviaciones estándar (DE) de la población normolipémica española (varones: 10,12mm; mujeres: 9,61mm)13.
Aislamiento y oxidación de LDLSe aisló LDL mediante ultracentrifugación secuencial a partir del plasma de un paciente HF homocigoto y se dializó a 4°C durante 24h con tampón fosfato salino (PBS). El contenido en proteína de LDL se determinó por el método de Bradford usando albúmina sérica bovina como calibrador. La LDL se diluyó en PBS a una concentración de 200mg/l para evitar su agregación y se oxidó mediante incubación con 8 pmol/l de CuCl2 durante 24h a 37°C. La reacción se detuvo añadiendo 1mmol/l de EDTA y posterior almacenamiento a 4°C. La oxidación de LDL se confirmó mediante tinción con negro de Sudán y electroforesis en gel de agarosa al 1% en tampón barbital y también por medida de dienos conjugados mediante espectrofotometría a una longitud de onda de 234nm.
Aislamiento y cultivo de monocitos/macrófagos humanosA cada sujeto seleccionado se le extrajeron 40ml de sangre periférica sobre heparina de litio (10 U/ml) como anticoagulante. Las células mononucleares se aislaron mediante centrifugación en gradiente de Ficoll-Paque (Amersham Bioscience Corp. Piscataway, NJ, EE.UU.), se lavaron 2 veces con RPMI-1640, sin L-glutamina y suplementado con antibióticos (100 U/ml de penicilina y 100μg/ml de estreptomicina) y se resuspendieron en RPMI-1640 completo (con GlutaMAX I y 25mM de tampón Hepes) suplementado con antibióticos (100 U/ml de penicilina y 100μg/ml de estreptomicina), 1% de aminoácidos no esenciales, 2% de piruvato sódico y 1% de suero humano autólogo decomplementado. La decomplementación del plasma se llevó a cabo por incubación en baño húmedo a 56°C durante 30min y posterior centrifugación a 3.000rpm durante 20min a 18°C. La viabilidad celular fue > 95% en todos los experimentos, lo que se comprobó mediante tinción con azul trypan. Las células se distribuyeron en 5 frascos de cultivo de 25cm2 a una densidad de 1,8 × 106 células/ml y se seleccionaron los monocitos mediante adhesión a los frascos durante 2h a 37°C y 5% de CO2. Tras este período de adhesión, se eliminaron las células no adheridas mediante varios lavados con PBS (sin calcio ni magnesio, pH = 7,4). Los monocitos se incubaron durante 24h con medio de cultivo RPMI-1640 completo con 10% de suero humano autólogo decomplementado. Después de este tiempo, los monocitos se lavaron con PBS y se diferenciaron a macrófagos en un medio libre de suero (Macrophage-Serum Free medium [Invitrogen Corp., Carlsbad, CA, EE.UU.]), incubando las células a 37°C durante 9 días en una atmósfera húmeda con un 5% de CO2.
Incubación con LDL oxidadaEn el día 9 se añadieron 50pg/ml de LDLox a 4 de los 5 frascos de cultivo durante 1, 3, 6 y 18h de incubación a 37°C. En el frasco restante se añadió PBS en lugar de LDLox, y se utilizó como control.
Aislamiento de ARN totalDespués de 10 días se retiró el medio de cultivo, se hicieron varios lavados de los macrófagos con PBS y se procedió a la extracción del ARN total mediante el kit RNeasy (Qiagen Inc., Valencia, CA, EE.UU.) siguiendo las instrucciones del fabricante. El ARN total se cuantificó mediante espectrofotometría a 260nm y se comprobó su pureza mediante la razón A260nm/ A280nm. La integridad del ARN se verificó mediante electroforesis en gel de agarosa al 1% y tinción con bromuro de etidio.
Síntesis de cADNDos μg de cada muestra de ARN total se trataron con 1 U de DNasa I (Ambion Corp., Austin, TX, EE.UU.), en tampón 20mM Tris–HCl, 50mM KCl, 2mM MgCl2 a 37°C durante 45min, seguido de la inactivación de la enzima a 65°C durante 10min, para eliminar el ADN genómico que pudiera quedar tras la extracción de ARN. Para sintetizar la primera hebra de cADN, se mezclaron 2μg del ARN tratado con DNAsa I con 150ng de hexámeros al azar (Invitrogen Corp., Carlsbad, CA, EE.UU.) e incubando a 65°C durante 5min para desnaturalizar las estructuras secundarias del ARN. Posteriormente, se añadió una mezcla de 0,5mM de cada dNTP, 50mM Tris–HCl (pH = 8,3), 75mM KCl, 3mM MgCl2, 10mM DTT, 40 U RNasa OUT y 200 U Super-Script III-RNasa H– (Invitrogen Corp., Carlsbad, CA, EE.UU.). La reacción, en un volumen final de 20μl, se mantuvo 10min a 25°C para facilitar la hibridación de los hexámeros al ARN y posteriormente se incubó durante 1h a 50°C, finalizando la reacción con la inactivación de la enzima a 70°C durante 15min.
RT-PCR en tiempo realLa PCR en tiempo real se llevó a cabo utilizando TaqMan Universal PCR Master Mix (dNTPs, tampón de reacción y AmpliTaq Gold DNA polimerasa) (Applied Biosystems, Foster City, CA, USA), cebadores sin marcar y sondas TaqMan-MGB marcadas con el fluoróforo FAM: Assay on Demand para IL-1β, Hs00174097_m1; CXCL3, Hs00171061_m1; PPARγ, Hs00234592_m1 IL-8, Hs00174103_m1; TNF-α, Hs00174128_m1; NF-κBIA, Hs00153283_m1 18srRNA, Hs099999901_s1; RPLP0, Hs99999902_m1 y HPRT1, Hs99999909_m1; y Assay by Design para triptasa α2, basado en la secuencia AF206665 (posición 320–653 pb), en un aparato ABI Prism 7000 HT Sequence Detection System.
El análisis de los resultados se realizó con el Sequence Detector Software (SDS) (Applied Biosystems, Foster City, CA, EE.UU.). Cada reacción se llevó a cabo por triplicado. El cambio de expresión génica se calculó por cuantificación relativa utilizando el método ΔCt. La expresión se normalizó con los genes constitutivos 18srRNA, RPLP0 y HPRT1 usando el método geNorm14,15.
Análisis estadísticoLos datos se presentan como media ± DE y como mediana (rango intercuartil). Se comprobó que las variables cuantitativas seguían una distribución normal mediante el test Shapiro-Wilks. El análisis de los datos lipídicos entre los grupos estudiados se realizó mediante el test ANOVA de un factor, y el análisis de la expresión génica mediante el test de la t de Student o el test de la U de Mann–Whitney, según correspondiese. La correlación entre el tamaño del tendón de Aquiles y la expresión génica se llevó a cabo mediante el test de Spearman. El análisis estadístico se realizó con el software Statistical Package for the Social Sciences (SPSS Inc.) version 11.0, y se consideró significación estadística para todos los análisis un valor de p ≤ 0,05.
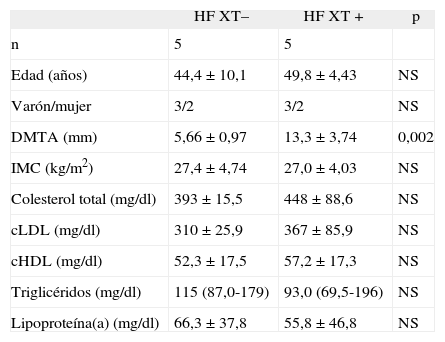
ResultadosDescripción de los sujetosLos datos lipídicos y antropométricos de los sujetos incluidos en este estudio se muestran en la tabla 1. Los pacientes HF XT+ presentaron un DMTA de 13,3 ± 3,74mm (media ± DE) y los pacientes HF XT– un DMTA de 5,66 ± 0,97mm (p = 0,002). No se encontró ninguna diferencia estadística entre el grupo de sujetos HF XT+ y el grupo HF XT– con respecto al sexo, colesterol total, cLDL, cHDL, triglicéridos o lipoproteína(a).
Datos lipídicos y antropométricos de los sujetos con hipercolesterolemia familiar del estudio, en función de la presencia o ausencia de xantomas tendinosos
| HF XT– | HF XT+ | p | |
| n | 5 | 5 | |
| Edad (años) | 44,4 ± 10,1 | 49,8 ± 4,43 | NS |
| Varón/mujer | 3/2 | 3/2 | NS |
| DMTA (mm) | 5,66 ± 0,97 | 13,3 ± 3,74 | 0,002 |
| IMC (kg/m2) | 27,4 ± 4,74 | 27,0 ± 4,03 | NS |
| Colesterol total (mg/dl) | 393 ± 15,5 | 448 ± 88,6 | NS |
| cLDL (mg/dl) | 310 ±25,9 | 367 ± 85,9 | NS |
| cHDL (mg/dl) | 52,3 ± 17,5 | 57,2 ± 17,3 | NS |
| Triglicéridos (mg/dl) | 115 (87,0-179) | 93,0 (69,5-196) | NS |
| Lipoproteína(a) (mg/dl) | 66,3 ± 37,8 | 55,8 ± 46,8 | NS |
cLDL: colesterol unido a lipoproteínas de baja densidad; cHDL: colesterol unido a lipoproteínas de alta densidad; DMTA: diámetro medio del tendón de Aquiles; HF: hipercolesterolemia familiar; IMC: índice de masa corporal; NS: no significativo; XT+: presencia de xantomas tendinosos; XT–: ausencia de xantomas tendinosos.
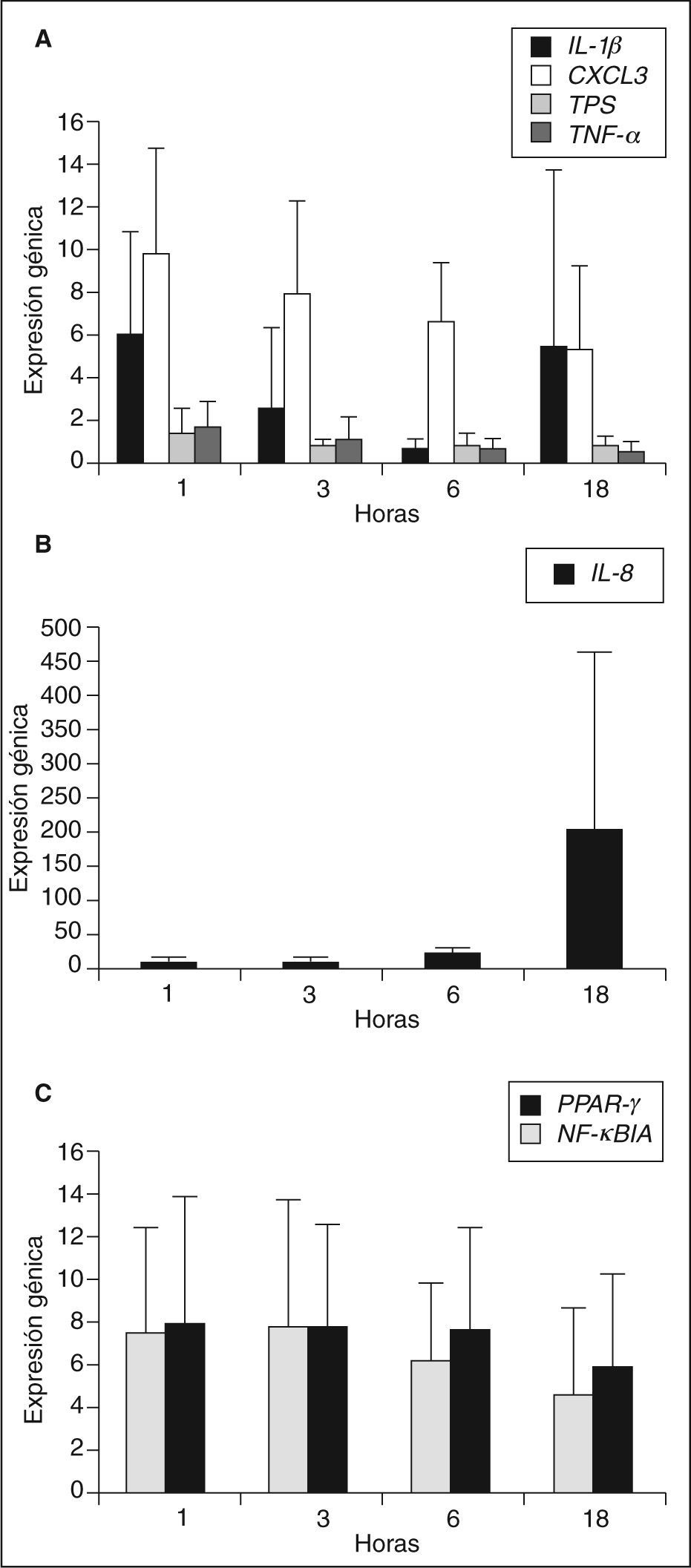
Mediante la técnica de PCR en tiempo real se analizó la expresión génica de diferentes moléculas inflamatorias en los macrófagos de los sujetos seleccionados incubados con LDLox durante diferentes tiempos de incubación: 1, 3, 6 y 18h. Los genes analizados fueron: el receptor nuclear PPARγ, el inhibidor del factor de transcripción NF-κB (NF-κBIA), las quimiocinas IL-8 y CXCL3, las citocinas IL-1β y TNF-α y la proteasa triptasa (TPS). Los genes analizados mostraron diferente patrón de expresión en función de las horas de incubación con LDLox, como se muestra en la figura 1. Los genes IL-1p, CXCL3, TPS y TNF-a alcanzaron su máxima expresión a tiempos cortos de incubación con LDLox (1 o 3h), IL-8 es el único gen de expresión tardía, alcanzando su máxima expresión a 18h de incubación con LDLox. No se encontró cambio de expresión en los genes PPARγ y NF-κBIA, manteniendo su expresión constante a lo largo del tratamiento con LDLox.
Expresión génica de los grupos HF XT+ y HF XT–, TNF-α. Panel B: IL-8. Panel C: PPAR-γ y NF-κBIA.")
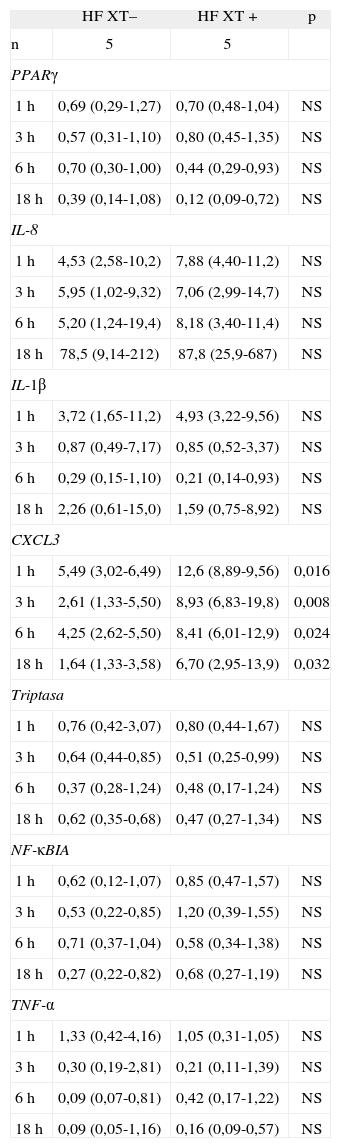
Para evaluar si había diferencias en la expresión de los genes analizados entre los sujetos HF con XT y los sujetos HF sin XT, se comparó dicha expresión génica entre ambos grupos de sujetos (tabla 2). Se encontró que el grupo de sujetos HF XT+ expresó más IL-8 tras 18h de incubación con LDLox que el grupo de sujetos HF XT-, aunque las diferencias no fueron significativas (87,8 [25,9-687] frente a 78,5 [9,14-212]). Sin embargo, se encontró que el gen TNF-α estaba menos expresado en el grupo HF XT+ tras 1h de incubación con LDLox (rango intercuartil: 0,31-1,05) que en el grupo HF XT– (rango intercuartil: 0,42-4,16), aunque estas diferencias tampoco alcanzaron significación estadística. No se encontraron diferencias en la expresión de los genes PPARγ TPS y NF-κBIA entre los grupos estudiados.
Datos de expresión génica de macrófagos de sujetos con hipercolesterolemia familiar estimulados con lipoproteínas de baja densidad oxidadas a diferentes tiempos de incubación, en función de la presencia o ausencia de xantomas tendinosos
| HF XT– | HF XT+ | p | |
| n | 5 | 5 | |
| PPARγ | |||
| 1h | 0,69 (0,29-1,27) | 0,70 (0,48-1,04) | NS |
| 3h | 0,57 (0,31-1,10) | 0,80 (0,45-1,35) | NS |
| 6h | 0,70 (0,30-1,00) | 0,44 (0,29-0,93) | NS |
| 18h | 0,39 (0,14-1,08) | 0,12 (0,09-0,72) | NS |
| IL-8 | |||
| 1h | 4,53 (2,58-10,2) | 7,88 (4,40-11,2) | NS |
| 3h | 5,95 (1,02-9,32) | 7,06 (2,99-14,7) | NS |
| 6h | 5,20 (1,24-19,4) | 8,18 (3,40-11,4) | NS |
| 18h | 78,5 (9,14-212) | 87,8 (25,9-687) | NS |
| IL-1β | |||
| 1h | 3,72 (1,65-11,2) | 4,93 (3,22-9,56) | NS |
| 3h | 0,87 (0,49-7,17) | 0,85 (0,52-3,37) | NS |
| 6h | 0,29 (0,15-1,10) | 0,21 (0,14-0,93) | NS |
| 18h | 2,26 (0,61-15,0) | 1,59 (0,75-8,92) | NS |
| CXCL3 | |||
| 1h | 5,49 (3,02-6,49) | 12,6 (8,89-9,56) | 0,016 |
| 3h | 2,61 (1,33-5,50) | 8,93 (6,83-19,8) | 0,008 |
| 6h | 4,25 (2,62-5,50) | 8,41 (6,01-12,9) | 0,024 |
| 18h | 1,64 (1,33-3,58) | 6,70 (2,95-13,9) | 0,032 |
| Triptasa | |||
| 1h | 0,76 (0,42-3,07) | 0,80 (0,44-1,67) | NS |
| 3h | 0,64 (0,44-0,85) | 0,51 (0,25-0,99) | NS |
| 6h | 0,37 (0,28-1,24) | 0,48 (0,17-1,24) | NS |
| 18h | 0,62 (0,35-0,68) | 0,47 (0,27-1,34) | NS |
| NF-κBIA | |||
| 1h | 0,62 (0,12-1,07) | 0,85 (0,47-1,57) | NS |
| 3h | 0,53 (0,22-0,85) | 1,20 (0,39-1,55) | NS |
| 6h | 0,71 (0,37-1,04) | 0,58 (0,34-1,38) | NS |
| 18h | 0,27 (0,22-0,82) | 0,68 (0,27-1,19) | NS |
| TNF-α | |||
| 1h | 1,33 (0,42-4,16) | 1,05 (0,31-1,05) | NS |
| 3h | 0,30 (0,19-2,81) | 0,21 (0,11-1,39) | NS |
| 6h | 0,09 (0,07-0,81) | 0,42 (0,17-1,22) | NS |
| 18h | 0,09 (0,05-1,16) | 0,16 (0,09-0,57) | NS |
HF: hipercolesterolemia familiar; NS: no significativo; XT+: presencia de xantomas tendinosos; XT-: ausencia de xantomas tendinosos. Datos expresados como mediana (rango intercuartílico).
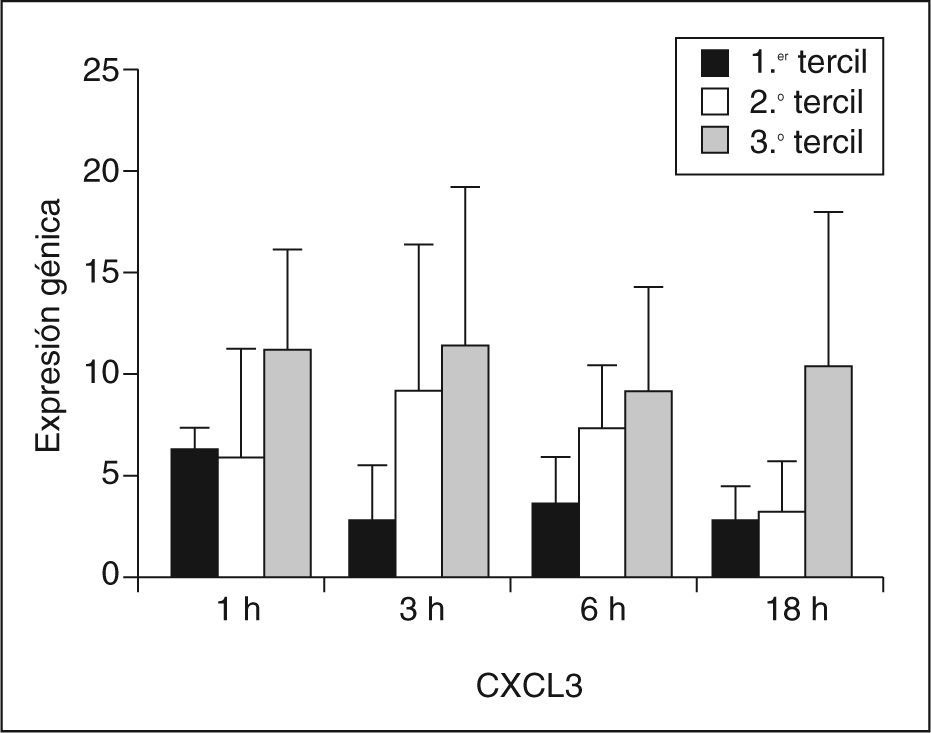
La expresión génica de CXCL3 fue significativamente mayor en el grupo de sujetos HF XT+ que en el grupo Hf XT– en todos los tiempos de incubación con LDLox. La mayor diferencia entre ambos grupos se encontró en el tiempo de 18h de incubación con LDLox, y la tasa de expresión de CXCL3 fue 4 veces mayor en el grupo Hf XT+ que en el grupo HF XT–. Para profundizar en la posible implicación de CXCL3 en la aparición de xantomas tendinosos en la HF, se analizó la expresión génica de CXCL3 en función del tamaño del tendón de Aquiles. Para ello, se dividieron los sujetos HF estudiados en terciles según el DMTA, y se analizó la expresión génica de CXCL3 en cada tercil y en cada tiempo de incubación con LDLox (fig. 2). El DMTA en el primer tercil fue de 5,13 ± 0,55mm, en el segundo tercil de 8,69 ± 2,67mm y en el tercer tercil de 14,8 ± 4,34mm. La menor expresión de CXCL3 a cualquier tiempo de incubación con LDLox tuvo lugar en el primer tercil de DMTA (1h: 6,16 ± 1,07; h: 2,79 ± 2,55; 6h: 3,66 ± 2,01; 18h: 2,61 ± 1,80), aumentando progresivamente en el segundo tercil (1h: 5,97 ± 5,20; 3h: 3,02 ± 7,30; 6h: 7,26 ± 3,11; 18h: 3,25 ± 2,39) y alcanzando su máxima expresión en el tercer tercil (1h: 11,26 ± 4,83; 3h: 11,34 ± 7,81; 6h: 8,96 ± 5,25; 18h: 10,41 ± 7,65). Se analizó la correlación entre la expresión génica de CXCL3 y el DMTA, encontrándose para cada tiempo de incubación con LDLox los siguientes coeficientes de correlación: 1h: R = 0,467, p = 0,174; 3h: R = 0,782, p = 0,008; 6h: R = 0,612, p = 0,060; 18h: R = 0,552, p = 0,098.
Discusión
La HF es una enfermedad monogénica causada por defectos en el gen del LDLR, cuya expresión clínica es altamente variable en términos de severidad, presencia de XT y aparición de enfermedad cardiovascular2. El mecanismo por el cual unos sujetos desarrollan XT y otros no, incluso compartiendo las mismas concentraciones de cLDL y la misma mutación en el gen del LDLR, es desconocido3. Trabajos previos de nuestro grupo de investigación han demostrado que los macrófagos de sujetos HF con XT tienen una respuesta inflamatoria diferente de la de los macrófagos de sujetos HF sin XT. Por ello, en el presente trabajo nos propusimos investigar la expresión génica de ciertas moléculas inflamatorias en relación con la aparición de los XT en la HF.
Nuestros resultados han demostrado que los macrófagos de sujetos HF con XT sobreexpresan el gen CXCL3 de manera significativa, así como el gen IL-8, aunque sin alcanzar significación estadística. CXCL3 es una quimiocina perteneciente a la familia GRO (growth-regulated oncogen), a la que pertenecen también otras 2 proteínas: CXCL1 y CXCL2. Estas proteínas actúan como mediadores en la inflamación, alergia e inmunidad16 y promueven la adhesión de los monocitos al endotelio vascular a través de la molécula VCAM-117. Trabajos previos han demostrado que la estimulación de monocitos humanos con LDLox aumenta la expresión de estas quimiocinas18,19. CXCL3 desencadena estas respuestas mediante unión al receptor CXCR216,20, proteína que se expresa en condiciones normales en monocitos circulantes y linfocitos T21,22, promueve quimiotaxis de monocitos, acumulación de macrófagos en la íntima, aumento de la adhesión de monocitos a células endoteliales23,24 y está implicada en la aterosclerosis avanzada25. Además, CXCR2 une otras quimiocinas del tipo C-X-C, como es el caso de IL-826. IL-8 comparte un 30-40% de homología con las 3 proteínas GRO16, es quimioatrayente para células T y células musculares lisas presentes en la placa de ateroma y fomenta la adhesión de monocitos a células endoteliales27. Además, se ha demostrado que el contenido intracelular de colesterol aumenta la expresión del gen IL-8 en macrófagos presentes en placas ateroscleróticas humanas28,29.
El hecho de que los macrófagos de sujetos HF con XT sobreexpresen los genes IL-8 y CXCL3 y, sobre todo, el hallazgo de que hay una fuerte correlación entre la expresión génica de CXCL3 y el DMTA, estaría de acuerdo con el papel de esta quimiocina en el reclutamiento, acumulación y adhesión de monocitos y macrófagos en el tendón para dar lugar a los xantomas y, por extensión, en la placa de ateroma. Por ello, IL-8 y, sobre todo, CXCL3, podrían tener un papel importante en la etiología de los xantomas mediante la activación de su receptor común CXCR2.
Nuestros resultados también mostraron que los macrófagos de sujetos HF sin XT tienden a sobreexpresar el gen TNF-α La citocina TNF-α se encuentra presente en placas ateroscleróticas humanas y de ratón30,31, se produce por diversos tipos celulares como macrófagos, monocitos o linfocitos T y es importante en la inmunidad innata y adaptativa, proliferación celular y procesos apoptóticos32. No se conoce con exactitud el papel de TNF-α en la aterosclerosis, algunos estudios le otorgan un papel proaterogénico33 y otros antiaterogénico34–37. Parece que el tipo de respuesta desencadenada por TNF-α depende del tipo celular y de la activación de receptores específicos; así, las células endoteliales desarrollarían una respuesta proaterogénica, mientras que el resultado en macrófagos y células musculares lisas sería antiaterogénico38. Estos últimos estudios apoyarían nuestros resultados, ya que la única citocina con mayor expresión en macrófagos de sujetos HF sin XT que en los de sujetos HF con XT fue TNF-α, lo que supondría para esta molécula un posible efecto protector frente al desarrollo de estos depósitos lipídicos. El mecanismo por el cual TNF-α ejerce su protección podría ser el propuesto por Gerbod-Giannone et al38, quienes demostraron que TNF-α induce la expresión del transportador ABCA1 a través de la ruta de NF-κB en macrófagos, aumentando el aclaramiento de colesterol hacia la apolipoproteína A-I y, por tanto, impidiendo que éste se deposite en las células espumosas.
En resumen, los macrófagos de sujetos HF con XT estimulados con LDLox presentan una sobreexpresión génica de las quimiocinas IL-8 y CXCL3, mientras que los macrófagos de sujetos HF sin XT presentan una tendencia a sobreexpresar el gen TNF-α. Estos resultados sugieren que las quimiocinas C-X-C podrían desempeñar un papel importante en la etiología de los xantomas, pudiendo ser CXCL3 un posible marcador de la aparición y desarrollo de estos depósitos lipídicos.
Los autores agradecen la colaboración de los pacientes en este estudio.
Este trabajo ha sido financiado gracias a los proyectos del Fondo de Investigación Sanitaria (PI03/1106, PI05/0247 y RD06/0014/0029 [RECAVA]) y Ministerio de Educación y Ciencia (SAF2005-07042).