




To explore the impact of obesity on the cardiac lipid profile in rats with diet-induced obesity, as well as to evaluate whether or not the specific changes in lipid species are associated with cardiac fibrosis.
MethodsMale Wistar rats were fed either a high-fat diet (HFD, 35% fat) or standard diet (3.5% fat) for 6 weeks. Cardiac lipids were analyzed using by liquid chromatography-tandem mass spectrometry.
ResultsHFD rats showed cardiac fibrosis and enhanced levels of cardiac superoxide anion (O2), HOMA index, adiposity, and plasma leptin, as well as a reduction in those of cardiac glucose transporter (GLUT 4), compared with control animals. Cardiac lipid profile analysis showed a significant increase in triglycerides, especially those enriched with palmitic, stearic, and arachidonic acid. An increase in levels of diacylglycerol (DAG) was also observed. No changes in cardiac levels of diacyl phosphatidylcholine, or even a reduction in total levels of diacyl phosphatidylethanolamine, diacyl phosphatidylinositol, and sphingomyelins (SM) was observed in HFD, as compared with control animals. After adjustment for other variables (oxidative stress, HOMA, cardiac hypertrophy), total levels of DAG were independent predictors of cardiac fibrosis while the levels of total SM were independent predictors of the cardiac levels of GLUT 4.
ConclusionsThese data suggest that obesity has a significant impact on cardiac lipid composition, although it does not modulate the different species in a similar manner. Nonetheless, these changes are likely to participate in the cardiac damage in the context of obesity, since total DAG levels can facilitate the development of cardiac fibrosis, and SM levels predict GLUT4 levels.
Explorar el impacto de la obesidad sobre el perfil lipídico cardiaco en ratas con obesidad inducida por dieta. Se evaluó, además, si estos cambios se asocian con fibrosis cardiaca.
MétodosRatas macho Wistar fueron alimentadas con una dieta con alto contenido en grasa (HFD; 35% grasa) o con una dieta estándar (3,5% grasa) durante 6 semanas. El análisis del lipidoma cardiaco se realizó mediante cromatografía líquida en tándem con espectrofotometría de masas.
ResultadosLas ratas HFD presentaron fibrosis cardiaca, estrés oxidativo y un aumento en el índice HOMA, adiposidad y los niveles circulantes de leptina así como una reducción en los niveles cardiacos del transportador de glucosa (GLUT 4) en comparación con las ratas controles. El análisis del lipidoma cardiaco mostró un aumento de los niveles de triglicéridos especialmente los que contenían ácido palmítico, esteárico o araquidónico, un incremento en los de diacilglicerol (DAG) aunque no cambios en los de diacilfosfatidilcolina y una reducción en los de diacilfosofatidiletanolamina, diacilfosfatidilinositol o de esfingomielinas (SM) en las ratas HFD en comparación con las control. Después del ajuste por otras variables (estrés oxidativo, hipertrofia cardiaca, índice HOMA), los niveles de DAG fueron predictores independientes de fibrosis cardiaca mientras que los de SM fueron de los de niveles de GLUT4.
ConclusionesLa obesidad ejerce un impacto importante sobre el lipidoma cardiaco. Estos cambios parecen participar en el daño cardiaco en el contexto de la obesidad ya que los niveles de DAG podrían facilitar el desarrollo de fibrosis miocárdica y los de SM los de GLUT 4.
Obesity has become a relevant health problem that is reaching epidemic proportions worldwide.1 Obese individuals show a higher risk of cardiovascular morbidity and mortality, which has been explained through chronic low-grade inflammation, increased oxidative stress and the metabolic alterations associated with obesity which can affect cardiac function.2 These conditions are linked to excess lipid accumulation not only in adipose tissue but also in non-adipose tissues, including the heart, which occurs when the storage capacity of adipocytes is exceeded.3
Lipids are important regulators of cardiac function, not only as the main energy substrate for cardiac mitochondrial oxidative metabolism but also by their role in membrane phospholipid remodeling, their activity as signaling molecules and ligands for nuclear receptors. However, increased myocardial lipid accumulation elicits an imbalance between cardiomyocyte fatty acid uptake and fatty acid oxidation,4 which can facilitate the accumulation of cardiotoxic metabolites that can exert deleterious effects on the myocardium.5 Clinical studies with proton magnetic resonance spectroscopy have demonstrated that increased intramyocardial triglyceride (TG) accumulation occurs before cardiac dysfunction in patients with type 2 diabetes mellitus and correlates with body mass index.6,7 These data supporting a link between cardiac lipid accumulation and myocardial dysfunction. Experimental studies8–11 have shown that the accumulation of some lipid species, including diacylglycerol (DAG), lyso phospholipids, acyl carnitines, ceramides and TGs, can affect cardiomyocyte function and lead to cardiac dysfunction. However, the potential mechanisms that link the lipid accumulation with the functional alterations are not well established.
Cardiac fibrosis is an important contributor to heart muscle dysfunction in obesity.12 The excessive extracellular matrix (ECM) deposition arises from the imbalance between ECM synthesis and degradation. An exacerbated deposition of ECM components can cause an aberrant remodeling that favors functional alterations because a reduced relaxing capability of the heart can increase its filling pressure and contribute to diastolic dysfunction. However, whether or not changes in lipid profile associated with obesity can affect cardiac fibrosis is still undetermined. Therefore, we explore the impact of obesity on the heart lipid profile through a lipidomic analysis in rats with diet-induced obesity as compared with controls. In addition, we evaluate whether the specific changes in lipid species could be associated with cardiac fibrosis.
MethodsAnimalsMale Wistar rats of 150g (Harlan Ibérica, Barcelona, Spain) received either a standard diet (3.5% fat; Harlan Teklad no. TD.2014; n=8) or a high-fat diet (HFD, 35% fat; Harlan Teklad no. TD.03307, Madison, WI, USA; n=8) for 6 weeks. The Animal Care and Use Committee of Universidad Complutense de Madrid approved all experimental procedures according to the Spanish Policy for Animal Protection RD53/2013, which meets the European Union Directive 2010/63/UE.
Body weight was measured once a week. Food and water intake were determined throughout the experimental period. Blood pressure (SBP) was estimated basally, at mid-study and end-of-study through the use of a tail-cuff plethysmograph (Narco Bio-Systems, Houston, TX, USA) in unrestrained animals. Serum and plasma were collected, fat pads were weighed, and heart was dissected for further analysis at the end of the experimental period. Adiposity index was calculated as sum of fat pad weight/(body weight-fat pad weight)×100).
Evaluation of cardiac structure and functionCardiac structure and function were evaluated by transthoracic echocardiography with a Philips CX50 (Philips, Netherlands) connected to a L12-3MHz linear transducer in rats anesthetized with isoflurane (2%; Esteve, Barcelona, Spain).
Measurements of left ventricular (LV) end-diastolic diameter, end-systolic diameter, interventricular septum (IVT) and posterior wall thickness (PWT) as well as calculations of left ventricular ejection fraction (EF) and LV systolic chamber function (pump function) were described elsewhere.13
Morphological and histological evaluationCardiac tissue samples were dehydrated, embedded in paraffin and cut into 4μm-thick sections. Sections were stained with picrosirius red in order to detect collagen fibers and viewed with polarized light under dark-field optics to detect the birefringence of collagen fibers. The area of cardiac interstitial fibrosis was identified as the ratio of interstitial fibrosis or collagen deposition to the total tissue area after excluding the vessel area from the region of interest. For each sample, 10–15 fields were analyzed with a 40× objective under transmitted light microscopy (Leica DM 2000; Leica AG, Germany). Myocytes (60–80 per animal) with visible nucleus and intact cellular membranes were chosen for determination of cross-sectional area in cardiac sections stained with hematoxylin and eosin.
Western blottingCardiac tissue lysates were separated by SDS-PAGE and transferred to 0.2μM nitrocellulose membranes (Bio-Rad Laboratories, Germany). Blots were incubated with antibodies against glucose transporter4 (GLUT4) (Santa Cruz Biotechnology Inc, Heidelberg, Germany). Bound antibodies were detected after incubation with an HRP-conjugated IgG and using the Super Signal West Dura Extended Duration Substrate (Thermo Fisher Scientific Inc, Waltham, MA, USA). Equal loading of protein in each lane was verified by β-actin (Sigma).
Detection of superoxide anion productionThe oxidative fluorescent dye dihydroethidium (DHE; Invitrogen, Grand Island, NY, USA) was used to evaluate superoxide anion (O2−) production. Cardiac and aorta tissue samples were embedded in tissue-freezing medium. 14-μm thick sections were then cut with a cryostat, placed onto glass microscope slides and washed briefly in Krebs-HEPES buffer (in mmol/L: NaCl 130, KCl 5.6, CaCl2 2, MgCl2 0.24, HEPES 8.3, glucose 11, pH 7.4). Slides were then incubated with DHE (5×10−3mmol/L) for 30min at 37°C in a light-protected humidified chamber. Slides were subsequently washed with warm phosphate-buffered. Cardiac images were viewed by fluorescent laser scanning microscope (40× objective in a Leica DMI 3000 microscope) (Ex561nm and Em610nm) using the same imaging settings in each case. Three separate histological sections and four different fields in each section per animal were quantified and averaged for each experimental condition. The mean fluorescence densities in the target region were calculated. Results are expressed as an n-fold increase over the values of the control group.
Lipidomic analysisMyocardial lipids were extracted and analyzed by ultrahigh performance liquid chromatography coupled to time-of-flight mass spectroscopy (UPLC-QToF-MS) using an Acquity UPLC System and a SYNAPT HDMS G2 (Waters, Manchester, UK) with electrospray ionization. Extraction of lipids was carried out from cardiac homogenates in methanol:chloroform mixture (1:2, v/v) and split into two aliquots. One aliquot was evaporated to dryness and the pellet re-suspended in acetone:2-propanol:ethanol (3:4:3, v/v/v) and used for TGs measurement. The other aliquot was evaporated to dryness and the pellet re-suspended in methanol:water (9:1, v/v/v) and used for phospholipids (PPLs) measurement. Extracts were kept at −80°C until analysis. Mass spectrometric analysis of TGs was performed in positive mode (ESI+) using the parameters that follow: capillary, 0.8kV; sampling cone, 15V; source temperature, 90°C; desolvation temperature, 280°C; cone gas, 40L/h; and desolvation gas, 700L/h. Data were acquired with the software MassLynx at a rate of 5 scans/s within the range 0–18min, and m/z 100–1200Da for the low-energy function and m/z 100–900Da for the high-energy function (MSE method, trap collision energy 30V). LC and MS methods were optimized using the commercial standards TG (18:2/18:2/18:2) and TG (16:0/16:0/16:0). These standards were also used to draw calibration curves for quantification. Mass spectrometric analysis of PPLs was fitted as follows: capillary, 0.9kV; sampling cone, 18V; source temperature, 90°C; desolvation temperature, 320°C; cone gas, 45L/h; and desolvation gas, 900L/h. Data were acquired with the software MassLynx at a rate of 5 scans/s within the range 0–12min and 100–1200Dam/z for the low-energy function, and 50–900Dam/z for the high-energy function (MSE method, trap collision energy 30V), with ionization in positive mode (ESI+) for detection of diacyl phosphatidylcholines (PCs), ceramides (CER) and sphingomyelins (SM), and with ionization in negative mode (ESI−) for detection of other phospholipids, which were diacyl phosphatidylethanolamine (PE), diacyl phosphatidylinositol (PI), diacyl phosphatidylglycerol (PG), and phosphatidic acids (PA). External commercial standards, namely PI (8:0/8:0), PG (14:0/14:0), PE (12:0/12:0), PC (10:0/10:0) and PA (14:0/14:0) were purchased from Cayman Chemical (Michigan, USA) and used for method optimization and quantification.
Up to three different chromatograms were manually checked for mass spectral peak identification where possible. Within each chromatographic point, m/z values with an intensity >=700 were also checked for it in order to afford a defined chromatographic peak (Extracted Ion Chromatogram, EIC); if positive, the elemental composition tool was then used to determine all the possible chemical compositions (CnHmOpNsPrSt) that were compatible with the isotopic distribution (M, M+1, M+2 and M+3 peaks) of a given m/z value. Using LipidMaps, Metlin, CheBI, LipidBank and KEGG databases, a certain elemental composition was examined for possible known compounds. Where possible, acyl chains were identified by data from the high-energy function (fragmentation). As well, specific fragments in the high energy function (MSE) were considered for identification, in particular m/z 184.07 for PCs and SMs in positive ionization mode.
Statistical analysisData are expressed as mean±SEM. Normality of distributions was verified by means of the Kolmogorov–Smirnov test. Data were analyzed using an unpaired Student's t-test to assess specific differences among groups using GraphPad Software Inc. (San Diego, CA, USA). Pearson correlation analysis was used to examine association among different variables. To find the factors associated with cardiac fibrosis or protein levels of GLUT4, the β-correlation coefficients (slope or mean difference, along with their 95% CIs) were obtained using a linear regression model. The predetermined significance level was P<0.05.
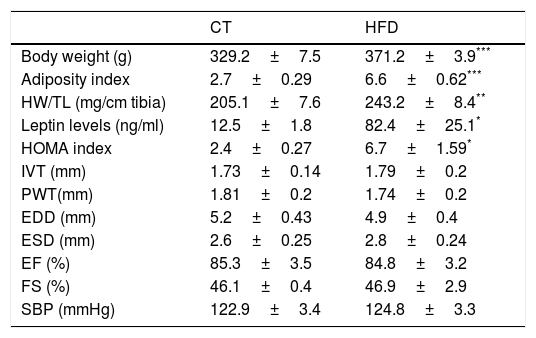
ResultsExperimental animals showed a progressive increase in body weight that was larger in the animals fed a HFD than in those fed a control diet. A similar increase was observed in adiposity index which was associated with plasma leptin levels (Tables 1 and 2). Our data demonstrated a significant increase in relative heart weight in obese animals as compared with the control group (Table 1). The HFD group showed also an increase in HOMA index (three-fold; Table 1), which suggests insulin resistance. The echocardiographic values for both structural features and left ventricular systolic function were similar in both groups (Table 1). No differences were found in systolic blood pressure at the end of the experiment between both groups (Table 1).
Body weight, adiposity, relative heart weight, leptin levels, HOMA index, echocardiographic parameters and systolic blood pressure in control rats and rats fed a high fat diet (HFD).
| CT | HFD | |
|---|---|---|
| Body weight (g) | 329.2±7.5 | 371.2±3.9*** |
| Adiposity index | 2.7±0.29 | 6.6±0.62*** |
| HW/TL (mg/cm tibia) | 205.1±7.6 | 243.2±8.4** |
| Leptin levels (ng/ml) | 12.5±1.8 | 82.4±25.1* |
| HOMA index | 2.4±0.27 | 6.7±1.59* |
| IVT (mm) | 1.73±0.14 | 1.79±0.2 |
| PWT(mm) | 1.81±0.2 | 1.74±0.2 |
| EDD (mm) | 5.2±0.43 | 4.9±0.4 |
| ESD (mm) | 2.6±0.25 | 2.8±0.24 |
| EF (%) | 85.3±3.5 | 84.8±3.2 |
| FS (%) | 46.1±0.4 | 46.9±2.9 |
| SBP (mmHg) | 122.9±3.4 | 124.8±3.3 |
HW: heart weight; TL: tibia length; HOMA index: the homeostasis model assessment; IVT: interventricular septum thickness; PWT: posterior wall thickness; EDD: end-diastolic diameter; ESD: end-systolic diameter; EF: ejection fraction; FS: fractional shortening; SBP: systolic blood pressure. Data values represent mean±SEM of 6 animals.
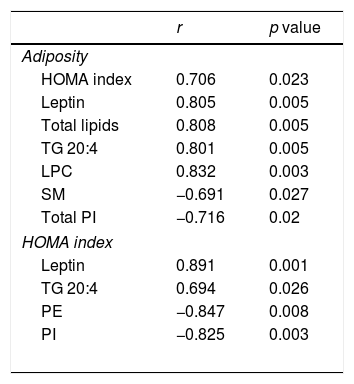
Associations found between adiposity index and HOMA Index and leptin plasma levels and cardiac lipid levels in control rats and rats fed a high fat diet (HFD).
| r | p value | |
|---|---|---|
| Adiposity | ||
| HOMA index | 0.706 | 0.023 |
| Leptin | 0.805 | 0.005 |
| Total lipids | 0.808 | 0.005 |
| TG 20:4 | 0.801 | 0.005 |
| LPC | 0.832 | 0.003 |
| SM | −0.691 | 0.027 |
| Total PI | −0.716 | 0.02 |
| HOMA index | ||
| Leptin | 0.891 | 0.001 |
| TG 20:4 | 0.694 | 0.026 |
| PE | −0.847 | 0.008 |
| PI | −0.825 | 0.003 |
HOMA index: the homeostasis model assessment; TG: Triglycerides; SM: sphingomyelins; PE: phosphatidylethanolamine; PI: phosphatidylinositol
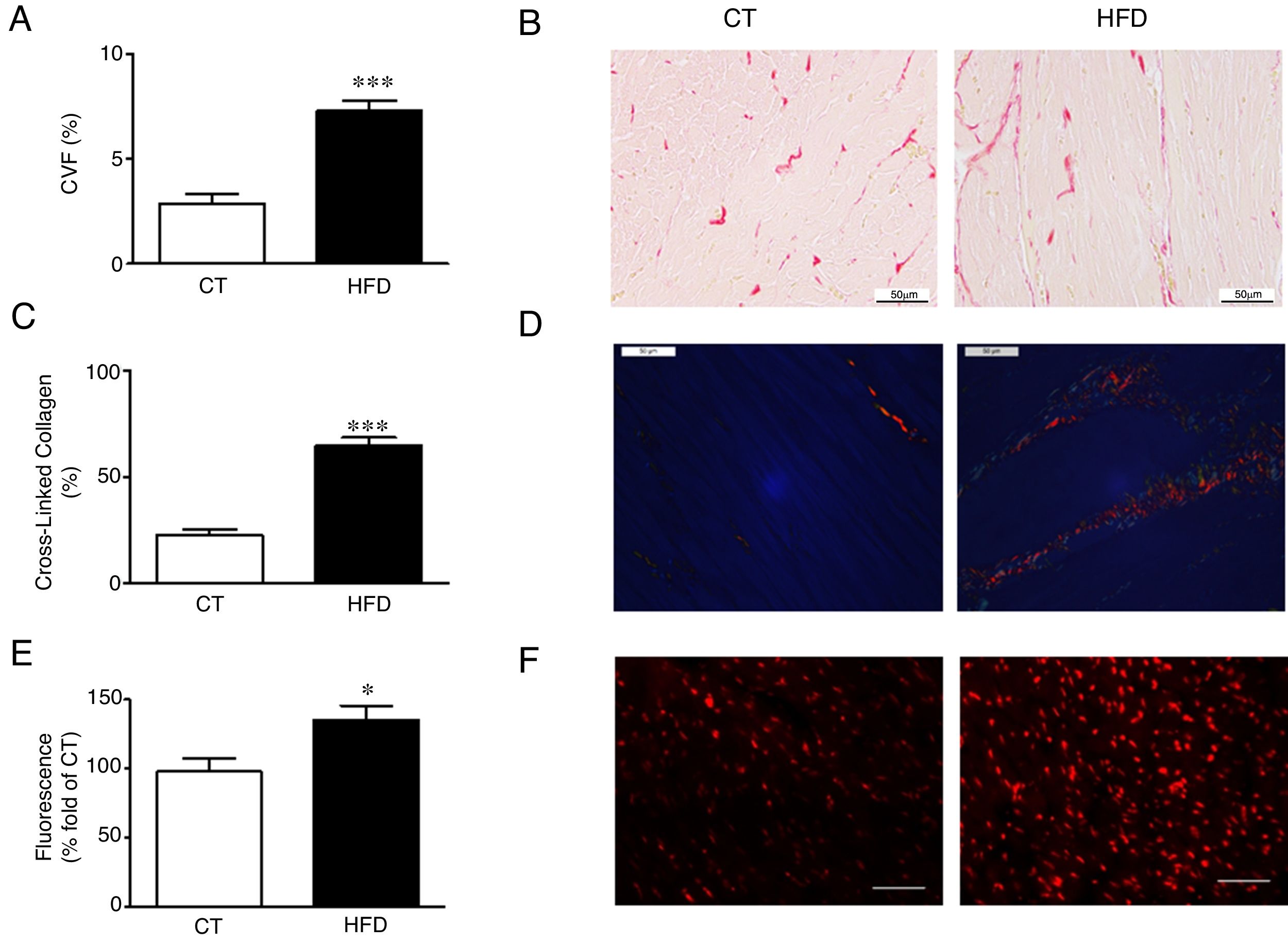
Diet-induced obese animals showed an increase (2.5 fold) in cardiac interstitial fibrosis in comparison to controls as demonstrated by the higher collagen volume fraction in the HFD group (Fig. 1A, B). This increase was mainly due to the cross-linked collagen, which increased 2.9 fold as compared with control group (Fig. 1C, D). The cardiovascular levels of O2− in obese animals were higher than those observed in control animals as suggested by the higher fluorescence intensity in myocardial tissue sections incubated with DHE (Fig. 1E, F) and were associated with collagen content (r=0.7808; p<0.001). No differences were observed in cardiac myocyte cross-sectional area among any of the groups (data not shown).
 Collagen volume fraction (CVF) and (B) representative microphotographs of myocardial sections staining with picrosirius red examined by light microscopy (magnification 40×) in heart from control rats (CT) and rats fed a high-fat diet (HFD). (C) Percentage of cross-linked collagen and (D) representative microphotographs of myocardial sections staining with picrosirius red examined by polarized light microscopy (magnification 40×) in heart from control rats (CT) and rats fed a high-fat diet (HFD). (E) Quantification of superoxide anions production in hearts from control rats (CT) and rats fed a high-fat diet (HFD) and (F) representative microphotographs of myocardial sections labeled with the oxidative dye hydroethidine by fluorescence microscopy (magnification 40×). Scale bar: 50μm. Values are mean±SEM of 8 animals. *p<0.05; ***p<0.001 vs control.")
Consequences of a high fat diet in the heart of rats. (A) Collagen volume fraction (CVF) and (B) representative microphotographs of myocardial sections staining with picrosirius red examined by light microscopy (magnification 40×) in heart from control rats (CT) and rats fed a high-fat diet (HFD). (C) Percentage of cross-linked collagen and (D) representative microphotographs of myocardial sections staining with picrosirius red examined by polarized light microscopy (magnification 40×) in heart from control rats (CT) and rats fed a high-fat diet (HFD). (E) Quantification of superoxide anions production in hearts from control rats (CT) and rats fed a high-fat diet (HFD) and (F) representative microphotographs of myocardial sections labeled with the oxidative dye hydroethidine by fluorescence microscopy (magnification 40×). Scale bar: 50μm. Values are mean±SEM of 8 animals. *p<0.05; ***p<0.001 vs control.
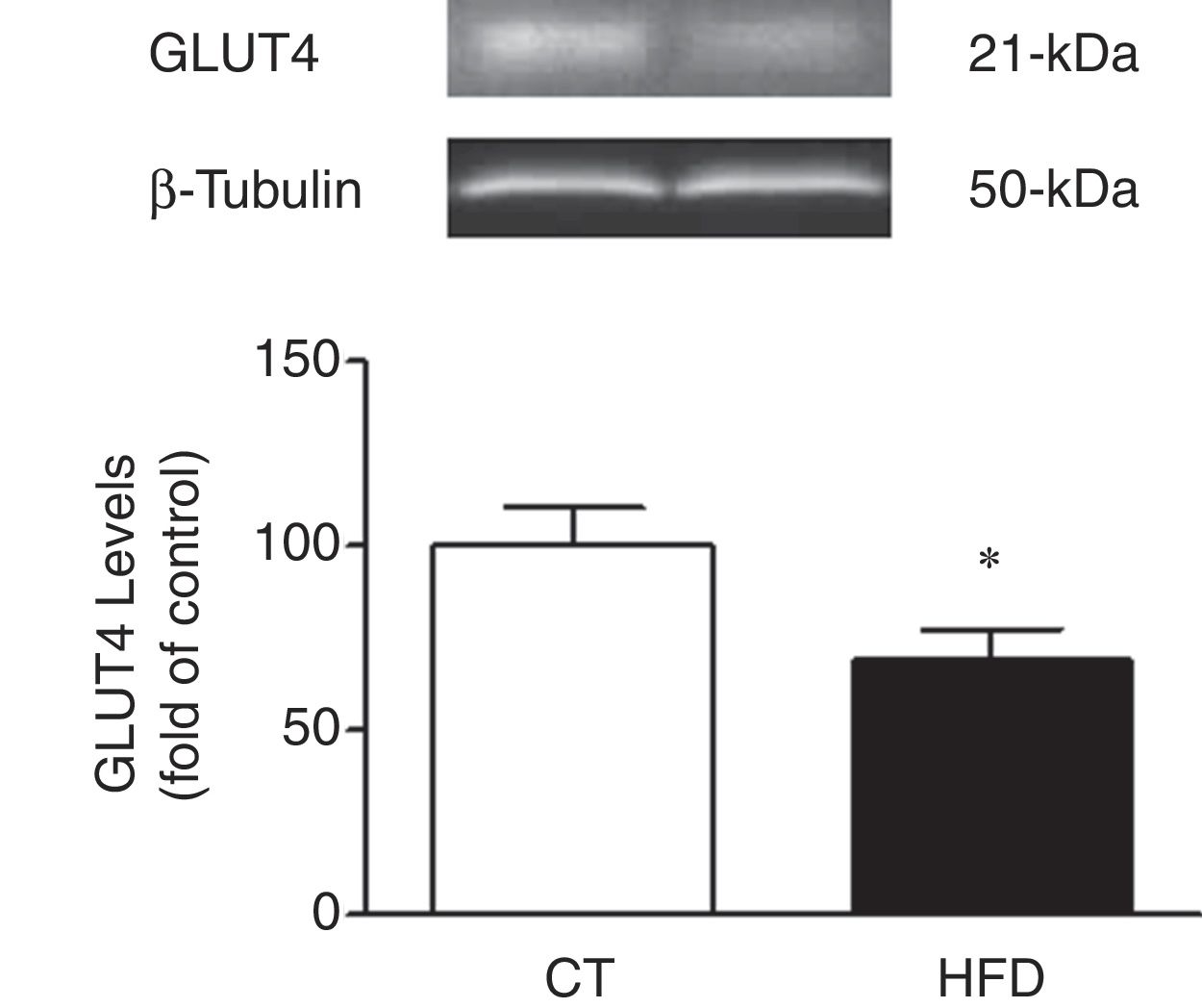
In agreement with the insulin resistance observed in obese animals, the cardiac protein levels of GLUT4 were reduced in HFD as compared to control rats supporting a reduction transport of glucose to the heart in obese animals (Fig. 2).

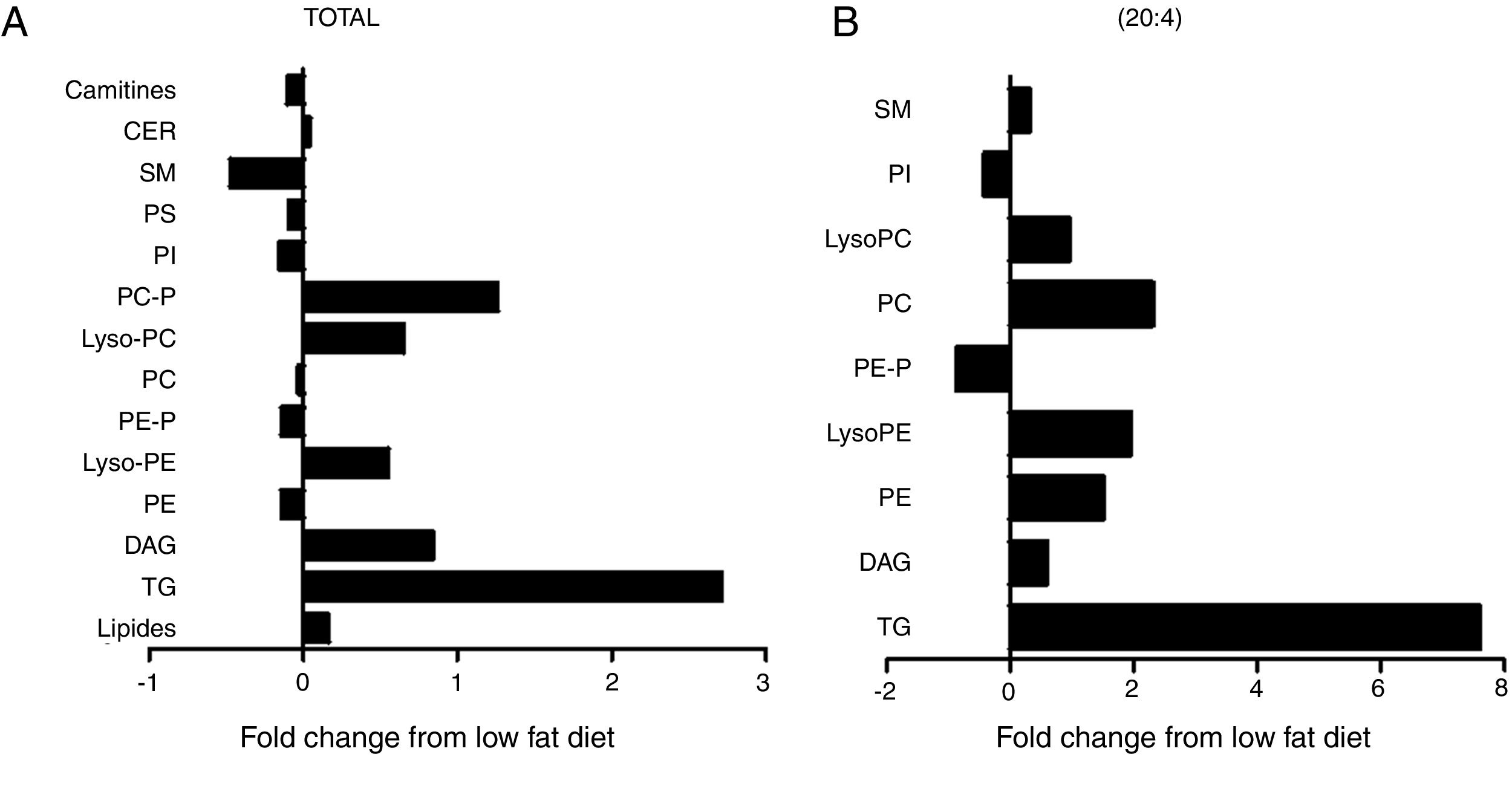
The cardiac lipidomic analysis detected overall 205 individual lipid subspecies over 12 different classes encompassing, among others, TG, DAG, SM, CER and PPLs. The analysis showed a relevant alteration in the cardiac lipid profile between HFD and control rats with changes identified in more than 50% of all species, although all lipid components are not affected in the same manner. The main lipid component was the PPLs, with PC and PE being the most abundant. Although no significant differences were observed between total levels of PC between both groups (Fig. 3A), a significant increase was observed in those species with 20:4 acyl chain (p<0.01; Fig. 3B) mainly PC (14:0/20:4), PC (18:0/20:4 and PC (18:1/20:4). By contrast, total cardiac PE levels were reduced (Fig. 3A; p<0.05), a fact mainly due to the reduction of the levels of PE containing palmitic acid (p<0.05), PE/PC ratio was consequently lower in HFD rats than in controls (2.3±0.04 vs 2.7±0.1; p<0.05; respectively). In addition, an increase was observed in lysoPE levels in HFD as compared with control animals (Fig. 3A). This increase was also observed in total lysoPC (LPC) species in obese rats versus normoweight animals (Fig. 3A; p<0.01). This increase was a consequence of the rise (p<0.01) observed in those animals of the major component, the LPC (20:4) (Fig. 3B). Both PI and diacyl phosphatidylserine (PS) species were also detected although at lower levels. However, the impact of the HFD was different in PI and PS: no changes in total levels of PS but a reduction (p<0.05; Fig. 3A) in total PI levels (Fig. 3A).
 levels of the different lipid species or (B) those containing arachidonic acid (20:4) as acyl chain in heart from rats fed a high-fat diet (HFD) as compared control rats (CT). TG: triglycerides; DAG: diacylglycerol; PE: diacyl phosphatidylethanolamine; LysoPE: lyso phosphatidylethanolamine; PE-P: plasmenil phosphatidylethanolamine; PC: diacyl phosphatidylcholine; LysoPC: lyso phosphatidylcholine PE-P: plasmenil phosphatidylcholine-P; PI: diacyl phosphatidylinositol; PS: diacyl phosphatidylserine; SM: sphingomyelins; CER: ceramides.")
Consequences of a high fat diet in the cardiac lipidome of rats. Changes in total (A) levels of the different lipid species or (B) those containing arachidonic acid (20:4) as acyl chain in heart from rats fed a high-fat diet (HFD) as compared control rats (CT). TG: triglycerides; DAG: diacylglycerol; PE: diacyl phosphatidylethanolamine; LysoPE: lyso phosphatidylethanolamine; PE-P: plasmenil phosphatidylethanolamine; PC: diacyl phosphatidylcholine; LysoPC: lyso phosphatidylcholine PE-P: plasmenil phosphatidylcholine-P; PI: diacyl phosphatidylinositol; PS: diacyl phosphatidylserine; SM: sphingomyelins; CER: ceramides.
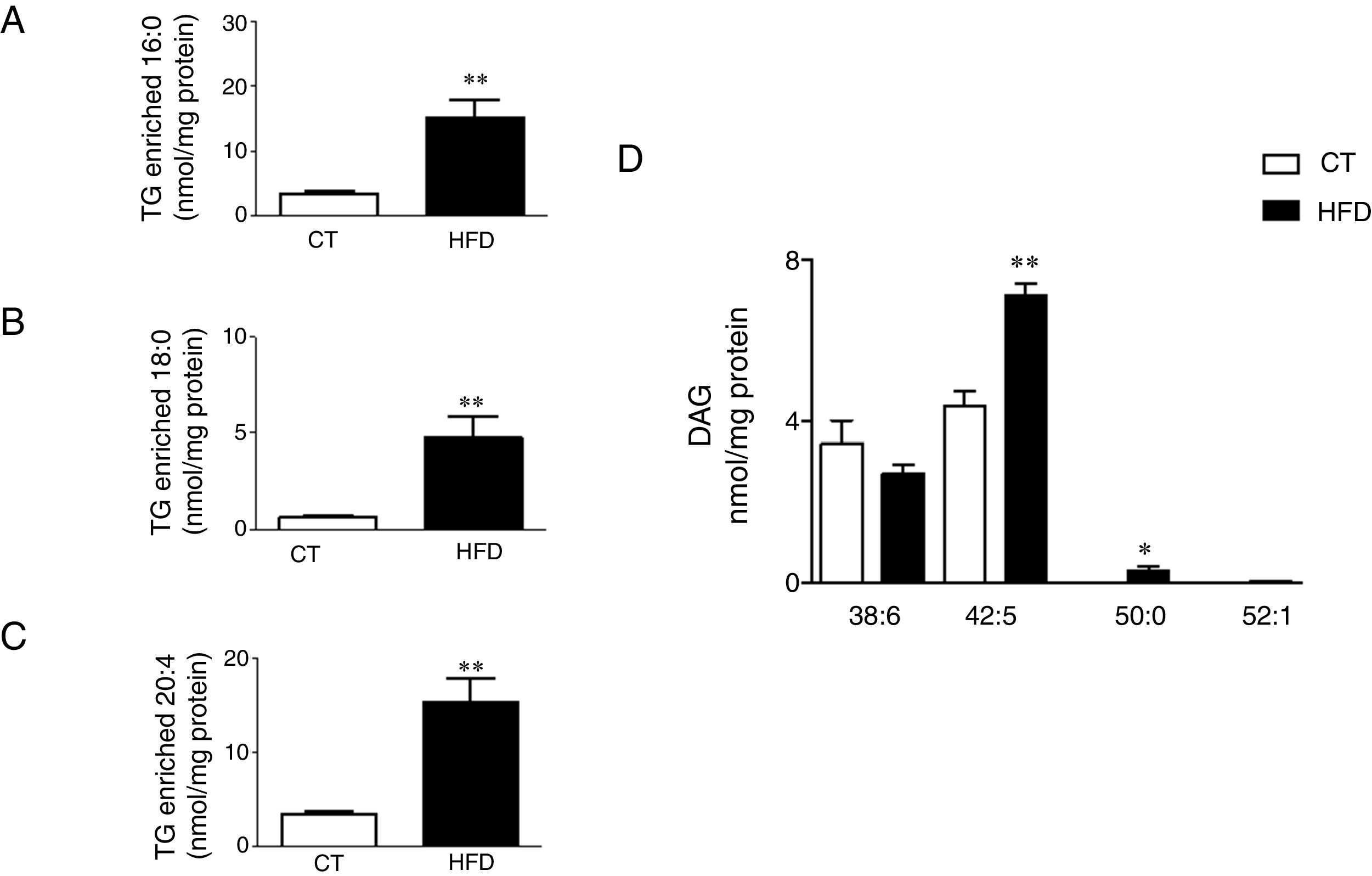
Total TG levels were increased (p<0.01; Fig. 3A) in HFD rats as compared with control animals by 2.7-fold. This increase was mainly due to those TG containing palmitic, stearic or arachidonic acids (Fig. 4A–C). In fact, 78% and 100%, respectively, of the detected TG species of each type were elevated, with the TG 50:1, 52:1 and 52:2 being the most abundant species (data not shown). The overall TG analysis showed that, as compared with control animals, the levels of those containing only saturated fatty acids were higher (3.8-fold increase) in HFD without significant changes in the levels of species containing only polyunsaturated or monounsaturated fatty acids. HFD rats also exhibited an increase of 7.6 fold in cardiac TG arachidonic acid (20:4) as acyl chain (p<0.01; Fig. 3B).
 species enriched with palmitic acid (A), stearic acid (B) and arachidonic acid (C) and of the diacylglicerol species (D) in the heart of rats fed a high-fat diet (HFD) and control rats (CT). Values are mean±SEM of 6–7 animals. *p<0.05; **p<0.01 vs control.")
Consequences of a high fat diet in the cardiac triglyceride and diacylglicerol species in rats. Levels of the triglyceride (TG) species enriched with palmitic acid (A), stearic acid (B) and arachidonic acid (C) and of the diacylglicerol species (D) in the heart of rats fed a high-fat diet (HFD) and control rats (CT). Values are mean±SEM of 6–7 animals. *p<0.05; **p<0.01 vs control.
Four species of DAG were detected in HFD group but only two of them were found in the heart of the control group (DAG 38:6; DG 42:5), which were the most abundant DAG species (Fig. 4D). Animals fed a HFD showed an increase in total DAG levels (0.85 folds; p<0.05; Fig. 3A), since 3 (DAG 50:0; DAG 52:1; DAG 42:5) of the 4 species detected were significantly elevated in HFD as compared with animals fed a control diet (Fig. 4D). In fact, the DAG containing C20:4 increased 0.6-fold (p<0.05; Fig. 3B) in the HFD group as compared with the control group. SM represented only around 5% of the cardiac lipids in control animals, and this content dropped to 1.4% in HFD animals (p<0.01) because of the drastic decrease (p<0.001) observed in the main SM specie detected in this study SM (d18:1/16:0). However, no reduction was observed in the levels of those SM with 20:4 (Fig. 3B). No differences were observed in levels of either sphingosine-1-phosphate (data not shown), carnitine or in the two detected species of ceramide between both groups (Fig. 3A). As shown in Table 2, adiposity index was correlated with levels of leptin, HOMA index, total lipids, arachidonic acid containing TGs and LPC levels. A negative correlation was found between adiposity and total levels of SM and PI. HOMA index was positively correlated with leptin levels and C20:4-containing TG content but a negative correlation was found of HOMA index to levels of PI and PE (Table 2).
In order to examine the relationship between cardiac lipid profile changes and cardiac fibrosis or cardiac levels of GLUT4, a linear regression analysis was performed. After adjustment for other variables which could affect cardiac fibrosis (HOMA index, ROS, and cardiac hypertrophy), it was found that total level of DAG could be considered an independent predictor of cardiac fibrosis (odds ratio 0.602; 95% CI, 0.012–0.683; p=0.04) whereas the level of total SM was shown to be an independent predictor of the cardiac level of GLUT 4 (odds ratio 28.18; 95% CI, 14.994–41.37; p=0.001).
DiscussionCardiac lipotoxicity has been associated with cardiac functional alterations in the context of obesity, which suggests that accumulation of lipids may exert a toxic effect on the myocardium.4,14 Even though cardiac lipotoxicity has been identified to accompany increases in TG levels, it is likely that the content and composition of other different lipid species are also altered and this imbalance can contribute to the cardiac damage associated with obesity, as well. In this study, we report a significant lipidomic remodeling in the heart of rats with diet-induced obesity relative to normally fed rats, the remodeling involving more than 50% of the lipid species detected in the heart. These changes, although affecting cardiac TG to a significant degree, also modify other species, including DAGs, SMs, PIs and LPCs, and this feature suggests that obesity has an effect on lipid content in the heart more than the one which had been foreseen. The observed changes in the obese animals seem to be relevant to the cardiac consequences of obesity since total DAG content seems to be a determinant factor for cardiac fibrosis, as does SM levels for the cardiac levels of GLUT 4; this suggests its potential role in the cardiac metabolic alterations in the context of obesity.
The data show that the increase in body weight observed in the model of diet-induced obesity (HFD animals) was associated with an important impact on cardiac lipid composition, with about 59% of the detected lipid species exhibiting a modified content. However, the change trends were species-specific, which resulted in only a slight increase in total lipids (16%) in HFD rats as compared with controls because of the increase in some species counteracted the decrease in other species. TGs were the main lipid class affected in regard to their content and profile, especially those containing stearic and palmitic acid were shown to have levels increased up to 8- and 4.5-fold, respectively, in HFD as compared with controls; this fact closely reflects the fatty acid composition of the diet. Indeed, significant correlations were found between adiposity, which was measured by the index of obesity and levels of either total TG or those enriched with arachidonic and palmitic acids. Such correlations support a close link between excess caloric intake and TG accumulation in the myocardium. These data are in agreement with those reported in previous studies, which demonstrated that cardiac or circulating TG levels were elevated in both obese patients and models of diet-induced obesity.3,15–18 More importantly, the changes observed in TG species in obese animals indicate that it is due to a profile more prone to cardiovascular complications. An imbalance due to either increased lipid uptake or decreased lipid oxidation has been proposed as the main cause underlying lipid accumulation. This idea is supported by studies in genetically modified mice, which affect a variety of components involved in lipid transport, storage, and metabolism.19–23 An enhanced lipid uptake has been suggested as a determinat factor involved in cardiac lipotoxicity in clinical studies in obese patients.24 In addition, it has been suggested that other lipid species could be facilitating heart TG accumulation. In this regard, Lim and Bodmer have shown that a reduction in PE levels through the modulation of the activity of the sterol regulatory element binding protein could facilitate lipogenesis in the heart of the model of easily-shocked Drosophila.25 Supporting this concept is the fact that our data showed a negative correlation between cardiac TG levels and those of PE (data not shown).
Although different mechanisms have been linked to TG accumulation and insulin resistance,26–28 some studies have suggested that TG are metabolically inactive since accumulation of TG has been observed in the muscle of insulin-sensitive women and athletes.29 Similarly, plasma TG is not associated with insulin resistance in overweight and obese patients.30 However, our data did not allow us to reach any conclusion regarding this feature because it is difficult to consider that changes in cardiac lipid species could have the ability to underlie the observed systemic insulin resistance. However, our data show that total SM levels, and especially the levels of SM (18:1/16:0), were independent predictors of GLUT4 cardiac levels, supporting previous data that suggest a positive role for SMs in insulin sensitivity in patients.31 Similarly, it has been shown that dietary SM improves metabolic complications associated with diet-induced obesity in mice.32
Cardiac interstitial fibrosis is a common feature in the context of obesity, which contributes to the pathogenesis of diastolic dysfunction.12,33 As previously reported,17,18,34 HFD animals show an increase in interstitial fibrosis, although no functional changes in cardiac function were observed, probably due to the relatively short time of evolution of obesity. The increase in interstitial fibrosis is mainly due to crosslinking collagen that is less prone to degradation. We have recently reported that the administration of an inhibitor of the activity of lysyl oxidase that catalyses the covalent cross-link of collagen and elastin fibers35 reduced cardiac fibrosis in rats with diet-induced obesity.34 A variety of factors has been involved in the development of cardiac fibrosis, including adipokines such as leptin. In fact, this role involved the activation of oxidative stress, engaging downstream events, which mediate the activation of PI3K/Akt pathway and, consequently, the production of end effectors. Such effectors include TGF-β, CTGF and galectin-3, which are mainly responsible for the final synthesis of ECM in cardiac myofibroblasts, the main factor responsible for fibrosis.36,37 Cardiac fibrosis can cause an aberrant remodeling that favors functional alterations, since a reduced relaxing capability of the heart can increase its filling pressure and contribute to diastolic dysfunction. The general concept that lipotoxicity can participate in cardiac fibrosis and functional alterations is widely accepted.19,38–42 However, the potential mechanism involved is not well established. The present data further extend this concept because it shows that the levels of DAG can predict those of cardiac interstitial fibrosis in rats. In agreement with this concept, it has been reported that the cardiac overexpression of cardiac-specific diacylglycerol kinase, an enzyme that negatively controls the cellular levels of DAG, reduces cardiac fibrosis and improves ventricular remodeling in mice with myocardial infarction, diabetes or aortic constriction.43–45
The potential underlying mechanisms are not well established but different data have linked DAG to cardiac lipotoxicity.10,46,47 DAGs are intracellular second messengers capable of increasing the activity of protein kinase C (PKC), which promotes cardiac fibrosis and heart failure48–51 through the activation of galectin-3 and oxidative stress.48,51 Therefore, activation of these factors which we have previously reported can participate in the cardiac fibrosis observed in rats fed a HFD18 and could be a potential mediator through which DAG can participate in cardiac fibrosis in the context of obesity. Supporting the role of DAG in cardiac fibrosis is the fact that overexpression of DAG kinase which modulates the activation of PKC, reduced cardiac fibrosis in different pathological conditions.43,44,52 Interestingly, changes in the DAG fatty acid composition result in translocation and activation of distinct PKC isoenzymes.53,54 Thus, it has been shown that 20:4ω-6 enriched DAG are more efficient that those with 20:5ω-3 or 22:6ω-3 in activating PKCδ, PKC¿ and PKCα. On the contrary, activation of PKCβI by DAG-containing arachidonic acid was significantly lower than that induced by the DAGs containing ω-3 PUFAs. Given that PKC isozymes play different roles (including opposing roles), in specific aspects of cardiac remodeling in HFD, the differential activation of PKC isoforms possibly leads to different pathophysiological effects. This phenomenon may be involved in the influence of ω-3 and ω-6 polyunsaturated fatty acids in health and disease. Thus, our findings that DAG containing 20:4ω-6 is increased in HFD group might be linked to an activation of PKC isotypes such as PKCδ, PKC¿ and PKCα. In agreement, it has been reported that these isoforms can mediate cardiac fibroblast proliferation and collagen production,55,56 and activation of PKCδ has been associated to increased cardiac damage.57 Moreover, it has been reported that the presence of cardiac DAG enriched with ω-3 is associated with a reduction of PKC¿ and PKCα translocation.54
Finally, it is worth mentioning that other observed changes could be relevant to different aspects of cardiac function that were not evaluated in the study, such as reduction in the ratio PE/PC. The levels of PC and PE are key regulators of membrane integrity as well as of its biophysical properties, such as curvature and rigidity, which influence protein functions. Thus, it has been recently reported that the activity of GLUT4 is controlled by membrane PPLs composition.58 The lipid components of the membrane can impact not only cell function but also mitochondrial function, since PE and PC are the predominant PPLS in mitochondrial membrane.59 Reductions in PI levels, which play important roles in lipid signaling, cell signaling and membrane trafficking, could also be relevant.
In summary, these data suggest that obesity exerts an important impact on cardiac lipid composition, although it does not modulate the different species in a similar manner. While an increase in TG and DAG-species was observed, PPLs, the main lipid component in the heart either does not change (PC, PS) or decrease (PE, PI). These changes can participate in cardiac damage in the context of obesity. Specifically, DAG can facilitate the development of cardiac fibrosis and SM levels can modulate cardiac GLUT4 levels. Consequently, the decrease in SM observed in HFD rats could facilitate the changes in the metabolic substrate use for myocardium that occurs in obesity. Although further work is warranted to better understand the entire spectrum of the cardiac functional consequences of the alterations on the lipid profile, the data provide an understanding of these changes and an insight into their underlying complexity.
Ethical disclosuresProtection of human and animal subjectsThe authors declare that the procedures followed were in accordance with the regulations of the relevant clinical research ethics committee and with those of the Code of Ethics of the World Medical Association (Declaration of Helsinki).
Confidentiality of dataThe authors declare that no patient data appear in this article.
Right to privacy and informed consentThe authors declare that no patient data appear in this article.
FundingThis work was supported by funds from the Fundación Española de Arteriosclerosis/Sociedad Española de Arteriosclerosis (Basic Research Award 2015), from Plan Estatal I+D+I 2013-2016: PI15/01060 and SAF2016-81063. The study was cofunded by Fondo Europeo de Desarrollo Regional (FEDER), a way to build Europe. GMR was supported by the Program (Pomoción de Empleo Juvenil) from Ministerio de Economia y Competitividad. BG and IG were supported by the FPI Program from the Government of Castilla y León (co-funded by FSE).
Conflict of interestsThe authors declare that they have no conflicts of interest.
We thank Avelina Hidalgo, Blanca Martínez, Virginia Peinado, and Roberto Cañadas.