







Distintos estudios epidemiológicos han encontrado una asociación entre la obesidad, la resistencia a la insulina, la dislipemia y la hipertensión con un mayor riesgo de desarrollo de la diabetes mellitus de tipo 2 (DM2) y la enfermedad cardiovascular. Este conjunto de procesos fisiopatológicos se ha agrupado con el nombre de síndrome metabólico. El progresivo y constante aumento del síndrome metabólico en países desarrollados ha impulsado la investigación de nuevas herramientas terapéuticas para el tratamiento de sus distintos componentes.
El receptor activado por proliferadores peroxisómicos g (PPARg) es un receptor nuclear que regula distintos procesos fisiológicos y celulares, además de presentar actividad antiinflamatoria independiente de lípidos. Es por ello que este receptor se ha convertido en una diana terapéutica muy atractiva para prevenir y controlar distintos componentes del síndrome metabólico. Las tiazolidinedionas (TZD) son ligandos sintéticos de PPARg que unen y activan el receptor y, en consecuencia, producen un incremento modesto de la masa de tejido adiposo subcutánea y mejoran la acción de la insulina. Es por ello que en pacientes con DM2 la administración de TZD mejora las anomalías lipídicas y la resistencia a la insulina asociadas a la obesidad. Más recientemente se ha abierto un nuevo campo en el desarrollo farmacológico de las TZD para su aplicación en la arteriosclerosis, pues se ha descrito que son capaces de corregir la disfunción endotelial en pacientes insulinorresistentes. Además de las clásicas TZD, distintos compuestos son capaces de activar PPARg y mejorar la sensibilidad a la insulina. Dentro de estos nuevos ligandos de PPARg se encuentran compuestos derivados de L-tirosina, derivados de los ácidos fenilacético y propiónico, y antiinflamatorios no esteroideos. No obstante, las TZD y otros compuestos que activan completamente PPARg presentan numerosos efectos secundarios no deseados debido principalmente a la amplia distribución tisular de PPARg, que dificulta su aplicación terapéutica. Esta problemática ha impulsado la búsqueda de nuevos compuestos que activen PPARg de manera selectiva en tejidos específicos, como son por ejemplo los moduladores selectivos de PPARg (SPPARM). En esta revisión se examina el potencial y la eficacia de distintos ligandos de PPARg para el tratamiento de los distintos trastornos patológicos asociados con el síndrome metabólico.
Receptor activado por proliferadores peroxisómicos g
Los receptores activados por proliferadores peroxisómicos (PPAR) son factores de transcripción hormonal nucleares activados por ligando. Actualmente se conocen 3 subtipos distintos de PPAR: PPARa (NR1C1), PPARb/d (NR1C2) y PPARg (NR1C3). Cada uno de ellos se caracteriza por presentar un patrón de expresión tisular diferencial, una sensibilidad y afinidad variables para distintos ligandos, y un reclutamiento de cofactores específico, que les confiere un perfil de actividad biológica divergente. Como norma general se puede afirmar que PPARa regula la oxidación y la captación de ácidos grasos, PPARg regula la homeostasis de la glucosa y el almacenamiento de ácidos grasos, mientras que PPARb/d regula el metabolismo de los ácidos grasos y la homeostasis lipídica.
Los PPAR comparten 4 dominios estructurales (fig. 1A)1-3:

Figura 1. A) Estructura general del gen PPAR* humano. PPARg consta de 4 regiones distintas: dominio aminoterminal A/B, que contiene la función de activación-1 independiente de ligando (AF-1); dominio C, con el dominio de unión a ADN (DBD); dominio D, implicado en la interacción con los distintos cofactores; domino C-terminal E/F, que contiene los dominios de unión a ligando (LBD) y de transactivación AF-2. Las formas PPAR*1 y PPAR*3 se traducen en una proteína idéntica (477 aminoácidos), mientras que PPAR*2 contiene 28 aminoácidos adicionales en su extremo N-terminal (505 aminoácidos). B) Mecanismo de activación de la transcripción génica mediada por PPARg. PPARg puede ser activado por múltiples ligandos naturales y sintéticos. Cuando se une al ligando, el LBD de PPARg experimenta una serie de cambios conformacionales que estimulan la disociación de correpresores y la formación de un heterodímero entre PPAR y el receptor de retinoide X (RXR). Este heterodímero PPARg/RXR es capaz de unirse a coactivadores específicos y promover la transcripción génica mediante el reconocimiento y unión a motivos específicos dentro del promotor del gen diana (PPRE).
1.Dominio aminoterminal A/B, que contiene la función de activación-1 (AF-1) independiente de ligando, responsable de la transactivación intrínseca del receptor.
2.Dominio C, con el dominio altamente conservado de unión a ADN (DBD).
3.Dominio D, implicado en la interacción con los distintos cofactores que controlan la actividad transcripcional.
4.Dominio C-terminal E/F. Este dominio E/F contiene los dominios de unión a ligando (LBD) y de transactivación AF-2, responsables de la actividad de transactivación del receptor dependiente de ligando.
El gen que codifica para PPARg en humanos se localiza en el cromosoma 3p254 y se transcribe en 3 distintos ARN mensajeros, denominados PPAR*1, PPAR*2 y PPAR*3, que difieren principalmente en su extremo 5'3,5. Curiosamente, PPAR*1 y PPAR*3 se traducen en una proteína idéntica, mientras que PPAR*2 codifica para una proteína distinta, con 28 aminoácidos adicionales en el extremo N-terminal6, que le confiere una especificidad tisular característica7. No obstante, ambas proteínas presentan idéntica actividad biológica3. PPAR*1 se expresa en el intestino grueso y en células hematopoyéticas, pero sobre todo en tejido adiposo blanco (TAB)1,8. PPAR*1 también es la forma predominante en el riñón, el hígado, el músculo, el páncreas y el intestino delgado, aunque su expresión es menor1,9. La isoforma PPAR*2 es casi exclusiva del TAB, aunque también se detecta su expresión en el tejido adiposo marrón (TAM) y el músculo esquelético10. Finalmente, PPAR*3 se ha detectado únicamente en el intestino delgado y en macrófagos5.
PPARg puede ser activado por múltiples ligandos, entre los que destacan los ácidos grasos mono y poliinsaturados y sus derivados eicosanoides (prostaglandinas y leucotrienos). No obstante, los ácidos grasos poliinsaturados (p. ej., los ácidos grasos esenciales g-linoleico, a-linolénico y araqui dónico) se consideran activadores débiles de PPARg11-13. El ligando más estudiado con respecto a su capacidad de activación de PPARg es el derivado de la prostaglandina D2, 15-deoxi-D12,14-prostaglandina J2 (15d-PGJ2), cuya potencia como agonista es relativamente elevada (2-5 mM)14. No obstante, se desconoce su relevancia en mamíferos, pues su concentración fisiológica raramente alcanza los valores necesarios para activar PPARg14,15. Sea cual sea el ligando activador unido, el LBD de PPARg experimenta una serie de cambios conformacionales que estimulan la disociación de correpresores y la formación de un heterodímero entre PPAR y otro receptor nuclear, el receptor de retinoide X (RXR o NR2B) (fig. 1B)2. Este heterodímero PPARg/RXR es capaz de unirse a coactivadores específicos y promover la transcripción génica mediante el reconocimiento y unión a motivos específicos dentro del promotor del gen diana. Estos motivos específicos se denominan elementos de respuesta a proliferadores peroxisómicos (PPRE)16,17, y consisten en 2 hexámeros nucleotídicos (5'-AGGTCA-3') separados por un único nucleótido (DR1) o, más raramente, 2 nucleótidos (DR2)3,9. Estos PPRE se han identificado en un gran número de genes regulados por PPAR17. La identidad de los cofactores reclutados por PPARg depende del tipo de ligando unido. Hay distintos coactivadores cuya actividad sobre PPARg se ha demostrado experimentalmente, y entre ellos destaca el coactivador del receptor esteroideo-1 (SRC-1), la proteína de unión a CREB (CBP)/p300 y el coactivador-1 de PPARg (PGC-1)1,2,18. Cabe destacar que ninguno de estos cofactores parece ser específico del subtipo PPARg. En ausencia de ligandos, PPARg interactúa con correpresores transcripcionales, como son N-CoR (correpresor del receptor nuclear) o SMRT (mediador-silenciador de receptores de retinoide y hormona tiroidea), y así silencian la transcripción del gen al cual están unidos13,18. Los PPAR también pueden regular negativamente la expresión génica de otros factores de transcripción, como es el caso del factor nuclear kB (NF-kB), mediante un proceso de antagonización dependiente de ligando pero independiente de PPRE9. Este fenómeno se conoce como transrepresión, y se cree que es el responsable de las propiedades antiinflamatorias de los PPAR9.
Funciones fisiopatológicas de PPARg
PPARg en la diferenciación y función adipocitaria
El papel principal del TAB es la síntesis y acumulación de lípidos a partir de ácidos grasos y triglicéridos (TG) plasmáticos, mientras que el TAM funciona como órgano termogénico. En ambos casos PPARg interviene de manera importante, y esto es consistente con el hecho que PPARg es el subtipo principal de PPAR expresado en tejido adiposo19 y que, además, sea en este tejido donde se encuentran los valores máximos de expresión de PPARg18. En realidad, PPARg es suficiente para inducir la diferenciación adipocitaria, pues la simple sobreexpresión ectópica de PPARg en fibroblastos o mioblastos es capaz de inducir la diferenciación de adipocitos in vitro20. Igualmente, el tratamiento de preadipocitos murinos con agonistas sintéticos de PPARg promueve su diferenciación a adipocitos maduros21, y la administración de TZD a ratas estimula la diferenciación de preadipocitos marrones y causa la hipertrofia del TAM21,22. PPARg también regula la homeostasis energética celular me-diante la estimulación de la expresión de las proteínas desacopladoras mitocondriales UCP-1, UCP-2 y UCP-320. Por ejemplo, UCP-1 es fuertemente inducida en TAB de ratones obesos KKA y después del tratamiento con ligandos de PPARg23. Paralelamente a la estimulación de la diferenciación adipocitaria, la activación de UCP-1 en TAB puede afectar el catabolismo lipídico y limitar la acumulación intracelular de TG, de manera similar a como sucede en el TAM. Además de su actividad estimuladora de la adipogénesis, PPARg desempeña un papel importante en la regulación del metabolismo lipídico en adipocitos maduros. Diferentes estudios han descrito un aumento mediado por PPARg de los genes que controlan la captación y el almacenaje de ácidos grasos (acilcoenzima A sintetasa [ACS], proteína de tejido adiposo 2 [aP2], proteína de unión a ácidos grasos [FABP], lipoproteína lipasa [LPL] y fosfoenolpiruvato carboxicinasa [PEPCK])7,20 y, por el contrario, la represión de genes que inducen la lipólisis y la liberación de ácidos grasos (receptor adrenérgico b3, leptina y factor de necrosis tumoral [TNF]-a). Por todo ello, la activación de PPARg incrementa el peso corporal en humanos, especialmente por la estimulación de la formación de depósitos de grasa subcutánea.
PPARg e insulinorresistencia
La resistencia a la insulina, que se define como la capacidad reducida de la insulina para actuar de manera efectiva en tejidos periféricos (músculo, tejido adiposo, hígado), se asocia frecuentemente con la obesidad visceral y suele conducir a un aumento de los valores plasmáticos de glucosa, ácidos grasos libres (AGL), TG y lipoproteínas ricas en TG (lipoproteínas de baja densidad pequeñas y densas [sdLDL] y lipoproteínas de muy baja densidad [VLDL])24. En sujetos obesos, los adipocitos se vuelven hipertrofiados y menos sensibles a la insulina y, en consecuencia, liberan más AGL en sangre, por que son resistentes a la acción antilipolítica de la insulina. Esto conllevará la acumulación de AGL en los adipocitos y la acumulación ectópica de grasa en el músculo esquelético y en el hígado18, donde provocará una mayor producción de VLDL y, por lo tanto, una mayor presencia de TG en plasma24. La acumulación de lípidos también inducirá insulinorresistencia debido a la estimulación de la gluconeogénesis hepática y a la reducción de la captación de glucosa por inhibición del principal transportador de éste, GLUT4, en el músculo esquelético25. Cuando los valores elevados de glucosa no se puedan compensar por la secreción de insulina, ocurrirá un fallo en las células b pancreáticas que acabará provocando insulinorresistencia y DM2.
En el cruce entre la obesidad y la resistencia a la insulina se encuentra PPARg. Se ha descrito que en ratones heterocigotos PPARg+/, con una deficiencia parcial de este receptor, se produce una protección parcial frente a la resistencia a la insulina inducida26. Otras evidencias han sugerido un papel insulino-sensibilizador para PPARg. Primero, la buena correlación encontrada entre la potencia in vivo de distintos agonistas de PPARg como sensibilizadores a insulina y su afinidad de unión al receptor in vitro27. Segundo, los agonistas de RXR, que activan el heterodímero PPARg/RXR, también mejoran la sensibilidad a insulina en ratones27. Tercero, se ha descrito una mutación en el gen PPAR*2 de ratón que incrementa la actividad de PPARg y que protege de la resistencia a la insulina asociada a la obesidad28, mientras que en ratones sin expresión de PPARg en tejido adiposo, músculo o hígado se produce una mayor predisposición al desarrollo de resistencia a la insulina18,29. Finalmente, estudios recientes realizados en humanos han demostrado que mutaciones en el gen de PPARg provocan resistencia grave a la insulina y síndrome metabólico30. Los individuos portadores del polimorfismo Pro12Ala en el gen PPAR*2, que reduce ligeramente la actividad de este subtipo de PPAR en el tejido adiposo, muestran un incremento de la sensibilidad a la insulina, una mejora en la homeostasis de la glucosa y un índice de masa corporal menor31. Se ha sugerido que en estos sujetos los adipocitos son menores y más sensibles a la insulina debido a la menor actividad de PPARg2.
La activación de PPARg aumenta el flujo neto de AGL hacia el tejido adiposo debido al aumento del número de adipocitos, la mayor generación de AGL como resultado de la estimulación de la actividad LPL32 y la inducción de PEPCK33 y glicerol cinasa34, que estimulan la utilización de precursores de la gluconeogénesis (fig. 2). PPARg también regula la expresión de genes que estimulan la captación y metabolismo de la glucosa en el tejido adiposo, como por ejemplo GLUT4 (fig. 2)35. Una mayor captación de glucosa en los adipocitos estimula la incorporación de AGL a TG, puesto que la glucosa puede ser convertida a glicerol-3-fosfato vía glucólisis8. Por otro lado, el tejido adiposo también actúa como órgano endocrino y secreta múltiples adipocitocinas, incluyendo leptina, resistina, adiponectina, interleucina-6 (IL-6), inhibidor del activador de plasminógeno-1 (PAI-1), TNF-a y ácidos grasos, entre otras. La masa de tejido adiposo determina parcialmente el nivel de secreción de estas adipocitocinas, que a su vez controlan la captación de glucosa mediada por insulina en el músculo36. En adipocitos disfuncionales, el patrón de secreción de adipocitocinas se encuentra alterado. La activación de PPARg en el tejido adiposo regula la secreción de adipocitocinas, pues se ha descrito que reduce la expresión de leptina37, resistina38, PAI-139, TNF-a40 e IL-641, mientras que induce la adiponectina, una adipocitocina que presenta actividad sensibilizadora a la insulina y mejora el metabolismo de la glucosa42. Aunque la represión de la leptina por parte de PPARg podría contribuir a la resistencia a la insulina, la mayoría de estudios in vivo han demostrado que la leptina incrementa la sensibilidad a la insulina43,44. Por otro lado, el TNF-a reduce la captación de glucosa dependiente de insulina y, por lo tanto, su inhibición podría contribuir a mejorar el control glucémico1.
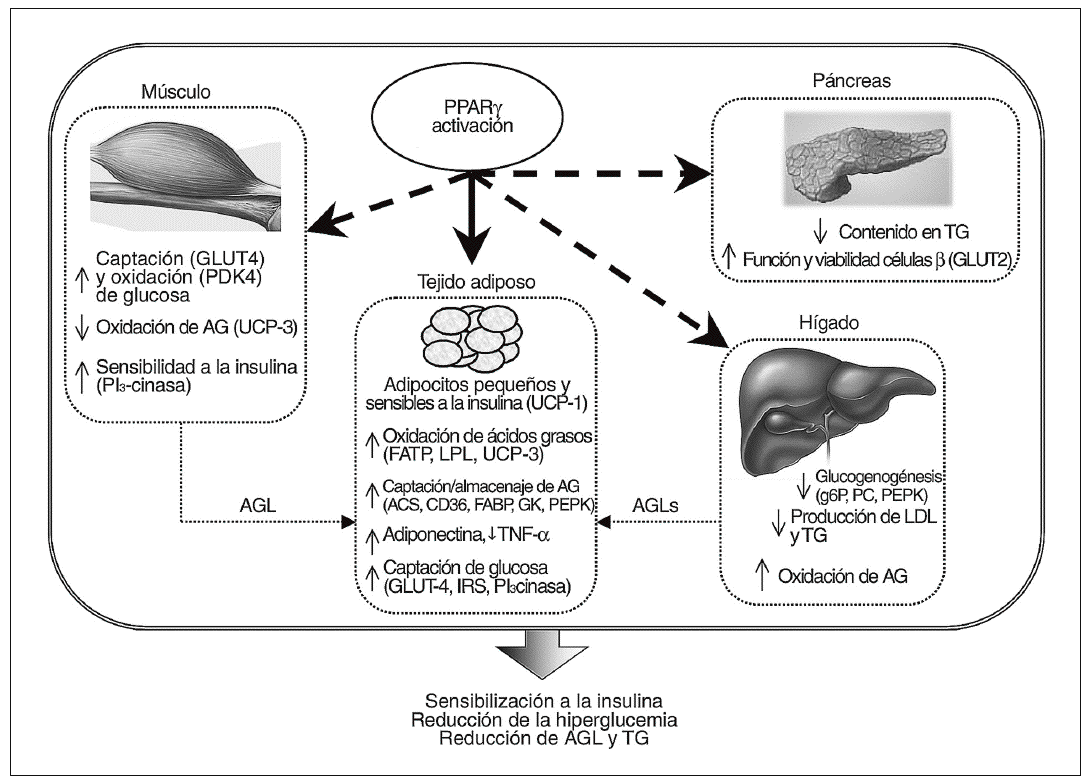
Figura 2. Representación esquemática de los mecanismos mediante los cuales la regulación de PPARg puede mejorar la sensibilidad a la insulina. La activación de PPARg en tejido adiposo estimula la diferenciación adipocitaria, aumentando el número de adipocitos pequeños más sensibles a la insulina y, reprime (*) o induce (*) vías metabólicas claves en distintos tejidos (páncreas, tejido adiposo, músculo esquelético e hígado). PPARg promueve la migración de ácidos grasos libres (AGL) desde el músculo y el hígado para su acumulación en el tejido adiposo, reduciendo así su liberación a la sangre. Una reducción en la expresión de factor de necrosis tumoral (TNF) a y una estimulación de la secreción de adiponectina aumenta aún más los efectos beneficiosos de la reducción de los AGL. Esto se produce concomitantemente a un aumento de la expresión de genes implicados en la captación de glucosa y la señalización de la insulina en tejido adiposo. Por otro lado, la activación de PPARg estimula la captación y oxidación de glucosa en el músculo, decreciendo además la producción hepática de glucosa (gluconeogénesis). Finalmente, PPARg parece prevenir la acumulación de triglicéridos (TG) en células b pancreáticas y, por lo tanto, su activación mejora la función de las células b. Los efectos de los ligandos de PPARg pueden ser directos (flecha continua) en tejido adiposo o directos/indirectos (flecha discontinua) en el músculo esquelético y el hígado. Los principales genes implicados en los efectos mediados por PPARg también se incluyen en la figura: ACS, acil CoA sintetasa; FABP, proteína de unión ácidos grasos; FATP, proteína transportadora de ácidos grasos; G6P, glucosa-6-fosfatasa; GK, glicerol cinasa; GLUT, transportador de glucosa; IRS, sustrato receptor de insulina; LPL, lipoproteína lipasa; PC, piruvato carboxilasa; PDK4, piruvato deshidrogenasa cinasa 4; PEPCK, fosfoenolpiruvato carboxicinasa; UCP-1 y UCP-3, proteínas desacopladoras 1 y 3, respectivamente.
No obstante, a pesar de la elevada expresión de PPARg en el tejido adiposo, éste sólo interviene en una pequeña fracción del aclarado de la glucosa dependiente de la insulina8. La expresión, aunque menor, de PPARg en otros tejidos esenciales para la homeostasis de la glucosa, como son el músculo esquelético, el hígado y las células b pancreáticas, sugieren otros mecanismos. De hecho, el músculo esquelético contribuye mayoritariamente (70%) a la utilización de la glucosa estimulada por insulina y es, consecuentemente, la principal diana de la resistencia a la insulina45. Probablemente, debido a ello, la supresión específica de PPARg en músculo de ratón produce resistencia a la insulina a causa de una alteración en la captación de glucosa en este tejido46. Con respecto al hígado, se ha observado que en distintos modelos animales de resistencia a la insulina y diabetes con esteatosis hepática la expresión de PPARg en hígado se encuentra marcadamente elevada7. Puesto que las TZD reducen la esteatosis hepática no alcohólica en humanos47, se ha sugerido que PPARg podría ser un mediador clave en la acumulación hepática de lípidos.
En resumen, diferentes mecanismos intervienen en la acción sensibilizadora a la insulina por parte de la activación de PPARg: estimulación de la captación de glucosa; inducción de la liberación de ácidos grasos de lipoproteínas ricas en TG; activación de genes implicados en la captación, síntesis y esterificación de ácidos grasos; inhibición de la expresión de TNF-a y leptina, e inducción de la adiponectina. Curiosamente, la acción antidiabética de los ligandos de PPARg en ratones obesos48 y en pacientes humanos49 se relaciona con la adipogénesis, la cual permite aumentar la capacidad de almacenamiento de lípidos y el restablecimiento de la secreción de adipocitocinas a valores normales.
PPARg y arteriosclerosis
La arteriosclerosis resulta de un complejo proceso inflamatorio crónico que implica la acumulación de lípidos en la pared arterial, con la consiguiente formación de células espumosas, migración de células mononucleadas, proliferación de células musculares lisas y formación de tejido fibroso9. Durante el proceso arteriosclerótico, los receptores de macrófagos tipo CD36 y el receptor scavenger de clase A (SR-A) unen lipoproteínas nativas y modificadas (p. ej., las LDL oxidadas), lo que conduce a la deposición intracelula;n de macrófagos en células espumosas50. La resistencia a la insulina contribuye a la disfunción endotelial, porque altera la vasodilatación mediada por óxido nítrico y aumenta la expresión de PAI-1 y la proliferación de células musculares lisas9,51. PPARg, cuya expresión es elevada en macrófagos, se ha asociado con el proceso de arteriosclerosis (fig. 3), pues se puede activar por distintos componentes de las lipoproteínas de baja densidad (LDL) oxidadas, como son los derivados metabólicos del ácido linolénico y los áci dos 9 y 13-hidroxioctadeca-9Z,11E-dienoico (9- y 13-HODE)17. La activación de PPARg con 15d-PGJ2 y otros agonistas estimula la expresión de CD36 en macrófagos50,52,53, y por lo tanto se esperaría que la activación de PPARg fuera proaterogénica por estimulación de la formación de células espumosas. No obstante, distintos estudios han demostrado lo contrario. Esta aparente paradoja se explicaría en parte por la inducción mediada por PPARg de la expresión del transportador ABCA1 (ATP-binding cassette transporter A1)54,55, implicado en el transporte reverso de colesterol y el flujo de colesterol desde los macrófagos, y a la supresión de la expresión del receptor SR-A56, que podrían compensar la inducción de la expresión de CD36.
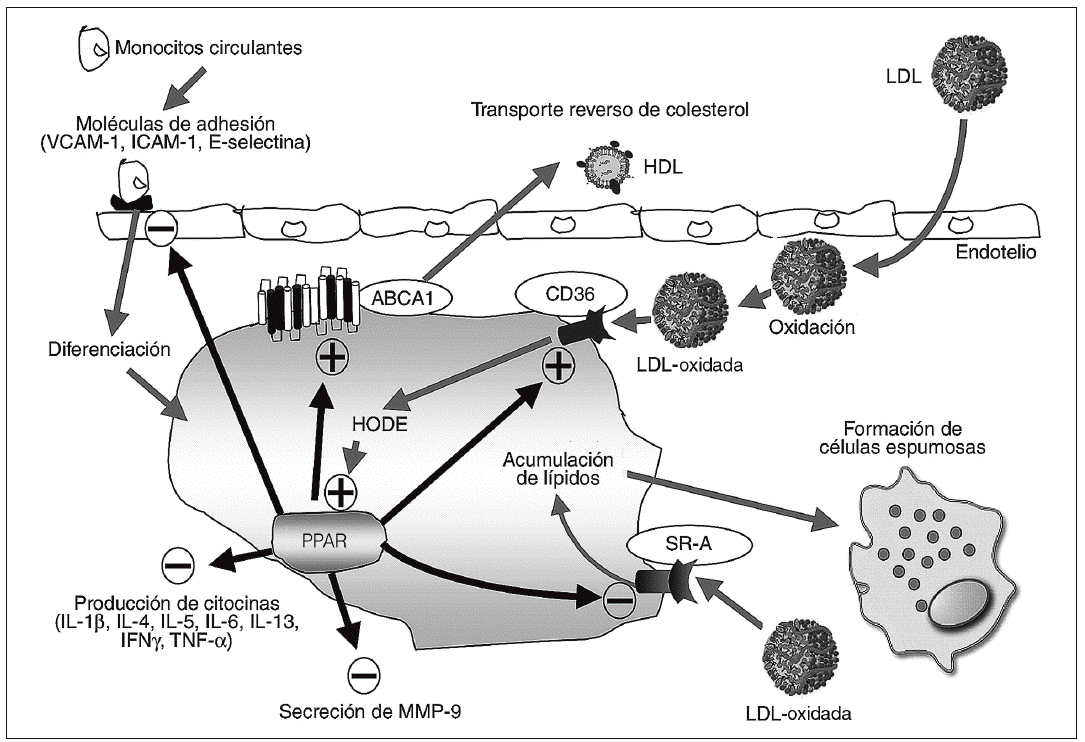
Figura 3. La activación de PPARg tiene efectos antiaterogénicos. PPARg puede ser activado por distintos componentes de las lipoproteínas de baja densidad (LDL) oxidadas, como son los derivados metabólicos del ácido linolénico 9 y 13-HODE. La activación de PPARg estimula la expresión de CD36 en los macrófagos. PPARg también induce la expresión del transportador ABCA1, implicado en el transporte reverso de colesterol y el flujo de colesterol desde los macrófagos, y suprime la expresión del receptor SR-A, disminuyendo por tanto la formación de células espumosas. La activación de PPARg también provoca efectos antiinflamatorios debido a su capacidad de inhibir la producción de citocinas proinflamatorias (interleucina [IL]-1b, IL-4, IL-5, IL-6, IL-13, IFNg y factor de necrosis tumoral [TNF-a]). También reduce la expresión de metaloproteasa de matriz (MMP), sintetasa de óxido nítrico inducible, ciclooxigenasa 2, proteína de atracción de monocitos-1 y moléculas de adhesión en distintos modelos, reduciendo así el reclutamiento de monocitos y macrófagos y mejorando la función endotelial.
La activación de PPARg también provoca efectos antiinflamatorios debido a su capacidad de inhibir la producción de citocinas proinflamatorias y de estimular la producción de adiponectina3. Estos efectos antiinflamatorios parecen ser independientes de su efecto sobre la acumulación lipídica. Esto es especialmente importante si se tiene en cuenta que PPARg, y particularmente PPAR*1, se expresa en muchas de las células presentes en la lesión arteriosclerótica, como son células endoteliales, musculares lisas y macrófagos. Distintos ligandos específicos de PPARg reducen la expresión de citocinas proinflamatorias (IL-1b, IL-4, IL-5, IL-6, IL-13, interferón g [IFN-g], TNF-a, metaloproteinasa de matriz 9 (MMP9), sintetasa de óxido nítrico inducible, ciclooxigenasa 2, proteína de atracción de monocitos-1 y moléculas de adhesión en distintos modelos in vivo y tipos celulares, reduciendo así el reclutamiento de monocitos y macrófagos y mejorando la función endotelial3,9,13,17,50,57,58. Algunos de los mecanismos propuestos mediante los cuales la activación de PPARg interferiría con el proceso inflamatorio serían la transrepresión de factores de transcripción tipo NF-kB, AP-1 y STAT-159, o la competición por los mismos coactivadores que estos factores de transcripción proinflamatorios3,18,50. También se ha sugerido que PPARg podría inhibir la respuesta inflamatoria mediada por la vía fosfatidilinositol 3-cinasa (PI3K)/Akt mediante la activación de la expresión de PTEN (phosphatase and tensin homologue deleted on chromosome 10)60. Otro mecanismo posible implicaría la inhibición de la ciclooxigenasa 2, enzima necesaria para la biosíntesis de las prostaglandinas desde el ácido araquidónico. En conjunto, estos datos sugieren que la activación de PPARg podría tener una aplicación terapéutica debido a sus efectos antiaterogénicos, ya sea evitando la formación de células espumosas o debido a sus efectos antiinflamatorios. No obstante, algunos datos contradicen estas propiedades beneficiosas y por lo tanto aún se requieren muchos más estudios para elucidar la viabilidad de esta propuesta61.
PPARg, hipertensión e hipertrofia cardíaca
Estudios en pacientes humanos con mutaciones dominantes negativas de PPARg han relacionado este receptor con la hipertensión grave2. Además, algunos agonistas de PPARg reducen la presión sanguínea en pacientes diabéticos y en distintos modelos animales de hipertensión62. El mecanismo responsable del efecto antihipertensivo de los agonistas de PPARg todavía se desconoce, aunque como posibilidades se han sugerido la inhibición de los canales vasculares de calcio, la regulación del sistema renina-angiotensina o la reducción de péptidos vasoactivos (endotelina)7,63. Por otro lado, también se ha sugerido un papel de PPARg en la regulación de la respuesta hipertrófica del corazón, pero existen ciertas divergencias al respecto64. Aunque PPARg se expresa poco en el corazón65, la hipertrofia cardíaca inducida por estrés mecánico, angiotensina II o fenilefrina en cardiomiocitos natales es inhibida por activadores de PPARg como la TZD troglitazona o 15d-PGJ2. La prevención de la actividad de NF-kB podría explicar parcialmente la inhibición de la hipertrofia cardíaca por parte de algunos agonistas de PPARg65.
Modulación farmacológica de PPARg
Los agonistas de PPARg pueden diferenciarse, según su actividad, en agonistas totales o agonistas parciales. Un agonista total es aquel compuesto que después de unirse a PPARg causa un reclutamiento completo de coactivadores, facilitando así una actividad transcripcional máxima. Por el contrario, un agonista parcial es capaz sólo de reclutar un subconjunto específico de coactivadores y, como resultado, únicamente activa parcialmente el receptor nuclear17. Por lo tanto, un agonista parcial también podrá presentar actividad antagonista sobre PPAR, pues podrá inhibir la actividad de un agonista total en función del contexto tisular y temporal del gen diana. Según su estructura química, los agonistas sintéticos de PPARg pueden clasificarse en TZD, derivados del ácido fenilacético o propiónico, derivados de L-tirosina y fármacos antiinflamatorios no esteroideos. En la actualidad también se están desarrollando agentes con capacidad para activar distintas isoformas de PPAR, conocidos como agonistas duales o pan-agonistas, según activen 2 o 3 subtipos distintos, respectivamente.
Tiazolidinedionas
El primer compuesto desarrollado del grupo de TZD fue la ciglitazona, que se vio que mejoraba la resistencia a la insulina y reducía la hiperglucemia sin estimular la secreción de insulina17. Posteriormente se han identificado otras TZD, como son la troglitazona, la rosiglitazona y la pioglitazona. Mediante ensayos in vitro se ha demostrado que las TZD se unen al PPARg con elevada afinidad y que son capaces de transactivar los genes diana a concentraciones del orden nanomolar52,66,67. La TZD clásica más potente es la rosiglitazona, pues se une al receptor con elevada afinidad (Kd ~40 nM)20. Las TZD se caracterizan por poseer la estructura tiazolidina-2-4-diona, pero difieren en sus cadenas laterales, que confieren el perfil farmacológico específico de cada compuesto68. La primera TZD introducida en la práctica clínica fue la troglita zona, aunque rápidamente se retiró del mercado debido a su hepatotoxicidad idiosincrásica rara pero grave8. Actualmente, sólo la rosiglitazona y la pioglitazona han sido aprobadas en Europa para el tratamiento de la DM2. Ambas se absorben rápidamente después de su administración por vía oral, y alcanzan su máxima concentración plasmática entre 1 h (rosiglitazona) y 2 h (pioglitazona) después de su administración69. En general, su farmacocinética no se altera después de la ingestión de alimentos, aunque en el caso de la pioglitazona el pico de máxima concentración se puede llegar a retrasar hasta las 4 h69. La rosiglitazona se metaboliza principalmente por los citocromos CYP2C9 y, sobre todo, CYP2C8, mientras que la pioglitazona se hidroliza y oxida por los citocromos CYP2C8 y CYP3A469. Aunque los metabolitos de la rosiglitazona son poco activos, los metabolitos de la pioglitazona conocidos como M-II y M-IV (derivados hidroxilados) y M-III (derivado cetónico) todavía son farmacológicamente activos69. La vida plasmática media depende de cada TZD, pero oscila entre las 4 h para la rosiglitazona y las 3-7 h o 16-24 h para la pioglitazona y sus metabolitos, respectivamente.
TZD en el síndrome metabólico y la resistencia a la insulina. Las TZD son agentes antidiabéticos orales que mejoran la sensibilidad a la insulina y la homeostasis de la glucosa. La ciglitazona redujo de manera importante la resistencia a la insulina en ratones obesos y diabéticos KKAy y mejoró sensiblemente la tolerancia a glucosa en ratas Zucker obesas y diabéticas (ZDF)17,70. Por otro lado, la ciglitazona y la rosiglitazona fueron capaces de moderar la resistencia a la insulina y de corregir la mayoría de alteraciones metabólicas asociadas con el envejecimiento en ratas Sprague-Dawley, a pesar de provocar un incremento de peso significativo71,72. Igualmente, la troglitazona y la rosiglitazona revirtieron la resistencia a la insulina y la DM2 inducidas mediante factores ambientales en distintos modelos murinos73-75.
El éxito de la terapia con TZD en pacientes de DM2 es contradictorio, puesto que al ser un factor estimulador de la adipogénesis se esperaría que la activación de PPARg incrementara la adiposidad y, en consecuencia, la captación de ácidos grasos por parte del tejido adiposo. Es por ello que uno de los efectos secundarios del tratamiento con TZD es un incremento de peso moderado18. Sin embargo, esta aparente paradoja se puede explicar parcialmente por los efectos colaterales de la activación de PPARg en el propio tejido adiposo y también en otros tejidos periféricos. En realidad, la activación de PPARg por las TZD favorece la redistribución del TAB, con una disminución de los depósitos viscerales en relación con los del área subcutánea. Probablemente esto es consecuencia de la mayor sensibilidad de los preadipocitos subcutáneos al efecto de las TZD7. Como PPARg se expresa predominantemente en tejido adiposo, este tejido debe desempeñar un papel importante en los efectos hipoglucemiantes de las TZD. Esto se ha demostrado en ratones sin tejido adiposo76 o sin actividad de PPARg en tejido adiposo77, que son refractarios a los efectos sensibilizadores a la insulina de las TZD. Las TZD mejoran la dislipemia diabética porque reducen los AGL y los TG plasmáticos mediante la estimulación de la captación de AGL por el adipocito, proveyendo así el sustrato necesario para la síntesis y acumulación de TG en el adipocito19. Numerosos genes implicados en el metabolismo lipídico son modulados por las TZD en adipocitos, como por ejemplo la proteína de tejido adiposo 2, la proteína transportadora de ácidos grasos, CD36 y el receptor de LDL oxidadas, que estimulan la captación de ácidos grasos; y LPL, ACS, PEPCK o glicerol cinasa, que promueven la hidrólisis de TG y la conversión de AGL a TG18,32-34. Además, las TZD inducen el coactivador PGC-1a, que promueve la biogénesis mitocondrial y, por tanto, estimulan la oxidación de ácidos grasos18. Las TZD también pueden modular directamente en adipocitos la expresión de genes implicados en la señalización de la insulina y la homeostasis de la glucosa, como son el sustrato 2 del receptor de insulina78, la subunidad p85 de PI3K79 y el transportador de glucosa dependiente de insulina GLUT435. El tratamiento con TZD provoca la formación de adipocitos más pequeños debido a un incremento de la diferenciación celular18 y una reducción en el número de adipocitos maduros, probablemente como consecuencia de la inducción de la apoptosis1, que causan globalmente un flujo de ácidos grasos hacia el tejido adiposo subcutáneo desde otros tejidos. Estos adipocitos pequeños, al ser más numerosos y más activos metabólicamente, responderán mejor a la insulina y producirán una respuesta más eficiente sobre la captación y posterior metabolismo de la glucosa en tejido adiposo17.
Otro mecanismo mediante el cual las TZD ejercen su influencia sobre el metabolismo de la glucosa es mediante la alteración del perfil de adipocitocinas secretadas. Las TZD reducen los valores de TNF-a, leptina y resistina2,38, mientras que inducen la secreción de adiponectina17. La adiponectina parece ser el mejor candidato para mediar los efectos sensibilizadores a la insulina de las TZD80, pues los valores de adiponectina plasmática se correlacionan directamente con la sensibilidad a la insulina e inversamente con la masa de tejido adiposo7, y además el tratamiento de ratones diabéticos con rosiglitazona normaliza los valores de adiponectina20. Por el contrario, la resistina, una hormona que altera la acción de la insulina y la tolerancia a la glucosa en el ratón, se reduce después de un tratamiento con TZD38.
Tradicionalmente se había considerado el incremento de sensibilidad a la insulina en el músculo y el hígado como un efecto colateral secundario de la reducción de los lípidos sistémicos mediante su acción en el tejido adiposo. No obstante, posteriormente se ha descrito que las TZD reducen la captación de AGL en el hígado y, como consecuencia, la tasa de gluconeogénesis también disminuye2. La administración de TZD también redujo la salida hepática de glucosa en la mayoría81,82, aunque no todos83, de los estudios clínicos realizados en humanos, y esto a pesar de los bajos valores de expresión de PPARg detectada en hígado humano7. Igualmente, también se han descrito efectos directos de las TZD sobre el tejido muscular, como por ejemplo en miotubos L684 y en células musculares esqueléticas humanas en cultivo in vitro85. El músculo esquelético, que es el sitio principal de utilización de glucosa, también parece ser la principal diana de acción de las TZD, aunque la expresión de PPARg es de 10 a 100 veces menor que en tejido adiposo17,86. En el músculo esquelético el mecanismo de sensibilización a la insulina de las TZD parece ser mediado por la inhibición de la síntesis de novo de ceramidas, así se evita la degradación de la proteína Akt87. Akt puede entonces ser fosforilada y activada por la insulina, promoviendo la captación y el metabolismo de la glucosa, además de reducir la oxidación de ácidos grasos mediante la reducción de la actividad de la proteincinasa dependiente de AMP (AMPK)87. La activación de PPARg por TZD (troglitazona y rosiglitazona) también puede proteger, e incluso restablecer, la función de las células b pancreáticas durante el desarrollo de la DM288-90, contribuyendo también a su efecto insulino-sensibilizador global. El tratamiento con TZD inhibe la acumulación intracelular de TG en células b pancreáticas mediante la estimulación de la oxidación de ácidos grasos, retrasando así la disfunción de las células b. Además, se ha encontrado un PPRE funcional en la región promotora de GLUT2, la proteína responsable del transporte de glucosa en células b1. Efectivamente, la rosiglitazona revirtió la alteración de la secreción de insulina en islotes pancráticos de ratas Sprague-Dawley derivada de la exposición a altas concentraciones de ácidos grasos91.
Eficacia clínica de las TZD. La troglitazona redujo la glucosa plasmática y la concentración de insulina en pacientes diabéticos, independientemente de la presencia de obesidad29,92. En estos pacientes se vio que la troglitazona reducía los valores de hemoglobina glucosilada, la glucosa plasmática e insulina en ayuno, los TG y AGL en plasma, además de incrementar la sensibilidad a la insulina93. La troglitazona también se mostró efectiva sobre la sensibilidad a la insulina y la protección contra la DM2 en mujeres con diabetes gestacional previa94. De manera similar, la pioglitazona disminuyó la hemoglobina glucosilada, la glucosa plasmática en ayuno, los AGL y los TG en pacientes con DM295-97. Igualmente, la rosiglitazona mejoró el metabolismo de la glucosa dependiente de insulina en pacientes con DM2 y redujo la concentración plasmática de glucosa y AGL y los valores hepáticos de TG98.
TZD en la arteriosclerosis e inflamación. La utilización de TZD para prevenir la arteriosclerosis en poblaciones de alto riesgo es controvertida, pues inicialmente se sugirió que la activación de PPARg podría estimular la formación de células espumosas. No obstante, diversos estudios apuntan a un efecto contrario. Recientemente se ha demostrado que la ciglitazona y la troglitazona reducen la acumulación de ésteres de colesterol en macrófagos cultivados in vitro99,100, contribuyendo así a reducir la formación de células espumosas. La troglitazona y la rosiglitazona también inducen la regresión de la placa y atenúan la arteriosclerosis entre un 20 y un 40% en modelos animales propensos a la enfermedad101-103. En estos animales, el contenido lipídico de los macrófagos después del tratamiento con TZD estaba compuesto principalmente de TG, a pesar de observarse un aumento de expresión de CD3617. La estimulación de la expresión de la carnitina palmitoiltransferasa I (CPT-I) podría explicar la reducción de los valores de ésteres de colesterol y de la formación de células espumosas, ya que CPT-I reduce la disponibilidad de ácidos grasos de cadena larga para su esterificación a colesterol y facilita el transporte reverso de colesterol100.
Aunque las TZD pueden mejorar la función endotelial mediante la supresión de la lipólisis, la mayoría de estudios también sugieren un efecto antia-terogénico alternativo e independiente de los cambios de insulina, glucosa o lípidos en circulación. En este sentido, las TZD ejercen múltiples efectos antiinflamatorios que podrían contribuir a su actividad antiaterogénica. La troglitazona y la rosiglitazona inhiben la proliferación, la hipertrofia y la migración de células musculares lisas vasculares104,105, y la formación de la placa en pacientes humanos54,106 y ratas ZDF107. Asimismo, las TZD inhiben la expresión de proteínas inflamatorias como IL-6, TNF-a, y MMP9 en macrófagos108 y reducen el PAI-1 en preadipocitos y adipocitos humanos109,110. No obstante, debido a las concentraciones elevadas de ligando necesarias para observar estos efectos se ha sugerido que éstos podrían ser mediados por mecanismos independientes de PPARg36. El bloqueo de la producción de citocinas por parte de la rosiglitazona en macrófagos derivados tanto de células embrionarias derivadas de PPARg+/+ como de PPARg/ apoyan esta teoría111. La inducción de la expresión de caveolina-1, un inhibidor de la sintetasa endotelial de óxido nítrico, por parte de la rosiglitazona112,113, o la inducción de enzimas con actividad antioxidante como la superóxido dismutasa de Cu/Zn por parte de la troglitazona y la pioglitazona114, podrían también explicar los efectos beneficiosos de la activación de PPARg sobre la arteriosclerosis. Igualmente, diversos estudios han demostrado que la troglitazona y la rosiglitazona reducen la generación de especies reactivas al oxígeno en pacientes obesos con o sin diabetes115,116, y que la rosiglitazona reduce de manera significativa la proteína C reactiva, un marcador de inflamación sistémica, MMP9 y E-selectina en pacientes no diabéticos con enfermedad coronaria arterial106 y en pacientes diabéticos117,118. Sea como sea, e independientemente de los mecanismos implicados, estudios retrospectivos en humanos119 y ensayos clínicos prospectivos en pacientes con DM2120 han demostrado un efecto ateroprotector para la activación de PPARg por parte de las TZD.
TZD en la hipertensión y la hipertrofia cardíaca. Las TZD reducen la presión sanguínea tanto en modelos murinos de hipertensión65 como en pacientes diabéticos121, sujetos insulinorresistentes29 o pacientes hipertensos122. Esta reducción en la presión sanguínea se correlaciona con una disminución en los valores de insulina, lo que sugiere que este efecto antihipertensivo de las TZD deriva del efecto indirecto de la insulina sobre la vasodilatación. Sin embargo, dado que las TZD también reducen la presión sanguínea en individuos hipertensos no diabéticos o en modelos animales hi pertensos sin resistencia a la insulina, se ha sugerido la presencia de algún mecanismo antihipertensivo independiente de la acción de la insulina2. Por ejemplo, las TZD pueden revertir la hipertensión por medio de la regulación transcripcional de genes implicados en el mantenimiento del tono vascular en células endoteliales, donde también se expresa PPARg como por ejemplo el péptido natriurético de tipo C, endotelina y PAI-1123. Estudios experimentales también han sugerido que la troglitazona podría reducir la presión sanguínea mediante el bloqueo de canales de calcio124.
Las TZD también se han asociado con la hipertrofia cardíaca en distintos modelos animales, aunque a concentraciones superiores a las recomendadas a nivel terapéutico64,125. Muchas de las consecuencias del tratamiento con TZD sobre la hipertrofia cardíaca dependen de la concentración de fármaco usada. Por ejemplo, se ha demostrado que la troglitazona a bajas concentraciones antagoniza la actividad de NF-kB en el corazón4, el cual se requiere para el crecimiento hipertrófico de los cardiomiocitos, mientras que a concentraciones elevadas induce la activación de NF-kB125. Por otro lado, la ciglitazona y la rosiglitazona no tienen ningún efecto sobre la expresión de genes implicados en el metabolismo lipídico cardíaco, como son ACS, FAT/CD36, CPT-I muscular, deshidrogenasas de acil-coenzima A de cadenas larga y media, proteínas desacopladoras 2 y 3 (UCP-2/3), ni tampoco sobre la tasa de oxidación de AGL125,126. Esto no haría sino reforzar los argumentos en contra de la regulación del metabolismo lipídico en el corazón mediado por PPARg. Existen algunos estudios que han demostrado que las TZD inhiben la hipertrofia cardíaca65,127. No obstante, también se ha sugerido que la troglitazona podría causar hipertrofia cardíaca a través de un ajuste en la expresión de genes implicados en la b-oxidación peroxisómica de ácidos grasos, en un mecanismo mediado por COUP-TF II125.
Efectos secundarios. A pesar de los beneficios clínicos de las TZD en el tratamiento de la DM2, su aplicación está limitada por sus numerosos efectos secundarios. Valores elevados de alanina aminotransferasa y alteraciones en la función mitocondrial hepática debidas a interacciones del fármaco con el citocromo CYP3A4 podrían contribuir a la toxicidad de estos compuestos1. Por ejemplo, la troglitazona fue retirada del mercado en el año 2000 debido a su hepatotoxicidad rara pero muy grave9. Independientemente de los efectos adversos detectados con la troglitazona, las otras TZD se consideran fármacos seguros y su uso aumenta progresivamente para tratar pacientes diabéticos25, pues parece que esta hepatotoxicidad no es un efecto de clase128. Algunos efectos específicos de clase observados en humanos, especialmente cuando se combinan con insulina, son edema sistémico, incremento de peso e infiltración de lípidos en la médula ósea2,8,9,69. El incremento de peso, aunque puede atribuirse en parte a la retención de líquidos, es consecuencia principalmente del aumento de la masa de tejido adiposo y suele ser proporcional a la afinidad para PPARg del ligalíquidos causada por las TZD, ésta deriva posiblemente de un aumento de la reabsorción renal de sodio y agua9,128,129. Tampoco se puede menospreciar la hipertrofia cardíaca observada en algunos pacientes después del tratamiento con TZD, que puede derivar en fallo cardíaco, o el aumento de la concentración de LDL en plasma provocada por la rosiglitazona al estimular la hidrólisis de lipoproteínas ricas en TG1. Aunque estos efectos adversos son poco frecuentes, sí son peligrosos, particularmente en individuos diabéticos con enfermedad cardiovascular2. Por esta razón se han dedicado esfuerzos ingentes en el diseño y el desarrollo de nuevos agonistas de PPARg con actividad sensibilizadora a la insulina pero sin los efectos adversos relacionados con las TZD.
Otros ligandos de PPARg
Ligandos de PPARg derivados de L-tirosina. El compuesto prototipo dentro de este grupo es GI262570, que une y activa PPARg con mayor afinidad y potencia (hasta 30-100 veces mayor) que las TZD, aunque su efecto antidiabético es equivalente. Un estudio clínico en fase II demostró que GI262570 tiene una potente actividad reductora de glucosa, y además es capaz de reducir los TG y aumentar el colesterol HDL en pacientes diabéticos2. Otros enantiómeros derivados del aminoácido L-tirosina y que unen PPARg humano a concentración nanomolar son GW1929 y GW7845130. GW7845 presenta un efecto similar a la troglitazona y la rosiglitazona con respecto a su efecto antiaterogénico en modelos animales101. Este grupo de compuestos se ha mostrado eficaz para el tratamiento de la diabetes y la insulinorresistencia en modelos animales, pero son muy potentes en la inducción de la adipogénesis y, por lo tanto, presentan los mismos efectos secundarios que las TZD131,132.
Derivados de los ácidos fenilacético y propiónico. Diversos derivados de los ácidos fenilacético y propiónico son capaces de unir y activar PPARg in vitro e in vivo: L-805645 (Ki 2 nM), L-165461 (Ki 15 nM), L-783483 (Ki 14 nM) y L-796449 (Ki 5 nM), este último un agonista PPARg/d dual muy potente128,133. Estos nuevos agonistas de PPARg fueron eficazmente utilizados como agentes insulino-sensibilizadores cuando se administraron oralmente a ratones diabéticos db/db, puesto que redujeron las concentraciones plasmáticas de glucosa y de TG133.
Fármacos antiinflamatorios no esteroideos. Algunos compuestos del subgrupo de fármacos antiinflamatorios no esteroideos (FAINE), entre los que destacan la indometacina, el diclofenaco, el fenoprofeno, el ibuprofeno y el ácido flufenámico, tienen actividad agonista sobre PPARg. Los FAINE son ligandos de baja afinidad que activan PPARg a concentraciones elevadas (50-500 mM), claramente superiores a las requeridas para la inhibición terapéutica de la ciclooxigenasa y la producción de citocinas inflamatorias134. En consecuencia, los FAINE son menos potente que las TZD con respecto a su capacidad de inducción de la diferenciación adipocitaria134 y, debido probablemente a ello, los FAINE también pueden actuar como antagonistas parciales de PPARg. Por ejemplo, la indometacina y el diclofenaco a concentraciones terapéuticas pueden inhibir la transactivación génica derivada de la inducción de PPARg mediada por TZD en células vasculares musculares lisas134. En conjunto, estos datos sugieren que los FAINE podrían actuar como agonistas de baja potencia o antagonistas parciales, dependiendo del contexto específico.
Otros compuestos. En la bibliografía hay numerosos ejemplos de extractos vegetales con actividad agonista de PPARg. Dos ejemplos clásicos son el extracto acuoso de la planta usada en medicina tradicional china Ginseng radix, que estimula la expresión de PPARg en tejido adiposo y es capaz de mejorar la hiperglucemia en ratones KKAy135, y el extracto orgánico de Momordica charantia, que activa PPARa y PPARg en células CHO-K1136. El saurufurano A, un furanoditerpeno acíclico obtenido de Saururus chinenesis, activa PPARg con una potencia similar a la ciglitazona137. También el ácido dehidrotrametenólico, un compuesto triterpenoide procedente de la planta medicinal china Poria cocos, estimula PPARg e incrementa la sensibilidad a la insulina en ratones db/db obesos138. Otros compuestos que son capaces de activar PPARg y promover la adipogénesis son la genisteína, derivado flavonoide de la planta de soja139, y los isoprenoides farnesol y geranilgeraniol, que activan tanto PPARa como PPARg140, y por lo tanto podrían regular la resistencia a la insulina y los valores plasmáticos de lípidos simultáneamente. El ácido ajulémico, un análogo sintético del ácido tetrahidrocannabinol-11-oico derivado de la planta Cannabis sativa, presenta actividad agonista selectiva sobre PPARg141 y modula profundamente el metabolismo lipídico142. Dado que es bien tolerado en humanos141,143, se ha sugerido que esta molécula podría ser útil para el tratamiento de DM2 así como de distintas enfermedades del metabolismo lipídico142. Finalmente, destaca el compuesto YM440, que une y activa PPARg y es capaz de reducir la glucosa e insulina plasmáticas en ratas ZDF sin inducir obesidad144. YM440 se une a PPARg con una afinidad similar a la pioglitazona (EC50 110 mM), aunque no induce la transactivación de PPARg o la diferenciación adipocitaria in vitro144. Esta actividad diferenciada de YM440 con respecto a las TZD podría ser consecuencia de los distintos cambios conformacionales producidos sobre la proteína PPARg, así como a un reclutamiento diferencial de cofactores. El principal órgano diana de YM440 es probablemente el hígado, a diferencia de otros agonistas de PPARg, pues mejora la resistencia a la insulina hepática pero no la periférica144.
Moduladores selectivos de PPARg
Los agonistas parciales modulan selectivamente la actividad PPARg mediante su unión diferencial a distintos cofactores, ya sea de manera cualitativa o cuantitativa, o bien evitando la fosforilación de PPARg. Los moduladores selectivos o parciales de PPARg (SPPARM) se han diseñado para mejorar la sensibilidad a la insulina sin provocar los efectos colaterales adversos no deseados. El primero de ellos que fue descrito es la acetamida GW0072, que presentaba actividad sensibilizadora a la insulina en ratas ZDF, pues reducía la insulina plasmática y los TG hasta valores comparables a los obtenidos con agonistas totales de PPARg, y sin causar tanto incremento de peso2. Esto último probablemente se debe a la menor capacidad de diferenciación adipocitaria in vivo de GW0072 en comparación con la rosiglitazona, pues la unión de GW0072 a PPARg reduce considerablemente el reclutamiento de CBP y SRC-1145. Además, GW0072 puede comportarse como antagonista total de la inducción adipocitaria inducida por la rosiglitazona145. Recientemente también se ha descrito otro potente SPPARM, FK614, un derivado benzimidazol146 que estimula la sensibilidad a la insulina en modelos animales de diabetes sin los efectos adversos derivados de un incremento de peso, una mayor adiposidad o la hipertrofia cardíaca147.
Otro ejemplo de agonista parcial de PPARg es FMOC-L-leucina (F-L-Leu), capaz de mejorar la sensibilidad a la insulina en ratones diabéticos ob/ob con menor potencia que las TZD148, y sin inducir ningún incremento de peso debido a su menor actividad estimuladora de la adipogénesis. Después de unirse a F-L-Leu, PPARg une el coactivador SRC-1, mientras que las TZD promueven la asociación con el factor intermediario de transcripción 2 (TIF-2), un regulador clave de la adiposidad, la termogénesis y la oxidación lipídica134. Este reclutamiento diferencial de cofactores contribuye probablemente a los efectos biológicos distintos entre F-L-Leu y las TZD. CLX-0921, un compuesto derivado del género Pterocarpus y usado antiguamente para tratar la diabetes en medicina indoamericana, es otro activador débil de PPARg. Resulta interesante observar que CLX-0921 presenta una actividad hipoglucemiante similar a la rosiglitazona en ratones C57/BL6, pero muestra una capacidad estimuladora de la adipogénesis 10 veces menor134. Esta reducción en la glucosa plasmática se acompaña de un incremento en la síntesis de glucógeno, característica no observada en otros agonistas de PPARg134. Además, CLX-0921 inhibe la biosíntesis de colesterol y de ácidos grasos134. La principal razón de esta actividad diferencial con respecto a las TZD podría residir otra vez en el perfil de reclutamiento de cofactores diferencial que presentan. Así, CLX-0921 recluta menos SRC-1 con respecto a la rosiglitazona134. El halofenato reduce los TG plasmáticos en pacientes dislipémicos, pero además también actúa como SPPARM, pues reduce la glucemia en pacientes de DM2 y es capaz de antagonizar la actividad de la rosiglitazona149. Este agonismo parcial se explicaría por el desplazamiento de los correpresores N-CoR y SMRT, combinado con un reclutamiento ineficaz de los coactivadores p300, CBP y TRAP220149. En ratas ZDF y ratones ob/ob, el halofenato mostró propiedades antidiabéticas sin incrementar el peso corporal y, además, mejoró la sensibilidad a la insulina en estudios a largo término149.
Otro grupo de compuestos con actividad selectiva sobre PPARg son los bloqueadores del receptor de angiotensina II (ARB), que se usan clínicamente como antihipertensivos. Los ARB ejercen su actividad antihipertensiva mediante el bloqueo del receptor de angiotensina II de tipo 1 (AT1) y la activación del receptor de angiotensina II de tipo 2 (AT2). Estudios recientes han sugerido una reducción del riesgo de diabetes de nuevo diagnóstico en pacientes tratados con ARB en comparación con otros fármacos antihipertensivos. Anteriormente, diversos estudios in vitro e in vivo ya habían sugerido esta capacidad para los ARB. Por ejemplo, el telmisartán y el irbesartán estimularon la diferenciación adipocitaria in vitro en células 3T3-L1150. In vivo, los ARB estimularon la captación de glucosa mediada por insulina en tejido muscular esquelético de ratas ZDF, conjuntamente con una mayor expresión de GLUT4 y una mejora en la tolerancia a la glucosa151. Resultados similares se obtuvieron cuando se administró olmesartán152. Se vio que el telmisartán también era capaz de modular selectivamente la expresión de genes diana claves de PPARg, como por ejemplo PEPCK y GLUT4153,154. En ratas alimentadas con dietas ricas en grasas e hidratos de carbono, el telmisartán redujo la hiperinsulinemia y provocó una reducción significativa de la glucosa y los TG plasmáticos, y atenuó significativamente el incremento de peso153. Finalmente, el telmisartán disminuyó la resistencia a la insulina derivada de la obesidad inducida con la dieta en ratones y, a diferencia de la pioglitazona, redujo el peso corporal debido a una disminución de la grasa corporal total155.
En humanos, el telmisartán mejoró la sensibilidad a la insulina en pacientes no diabéticos hipertensos156, y también mejoró el metabolismo de la glucosa y la función de las células b en sujetos insulinorresistentes157. No obstante, la adipogénesis en humanos depende de cada ARB. Mientras que el eprosartán no presentó actividad estimuladora de la adipogénesis ni activación de PPARg y el losartán sólo indujo la adipogénesis a dosis muy elevadas (100 mM), el irbesartán y el telmisartán eran efectivos a bajas concentraciones (1 y 0,1 mM, respectivamente)158. Las diferencias de actividad entre ARB y TZD probablemente dependen del reclutamiento específico de cofactores. Así, por ejemplo, la liberación del correpresor NCoR es menos acusada con ARB que con TZD, lo que explica su menor adipogénesis, mientras que el reclutamiento de TIF-2, coactivador implicado en la estimulación de la adipogénesis y de la lipogénesis por parte de PPARg, está reducida159. Los ARB presentan una potencia de activación de PPARg dependiente de la dosis: el telmisartán tiene una EC50 = 5,02 mM, que es similar al de la pioglitazona (EC50 = 1,5 mM); el irbesartán EC50 = 26,97 mM, y el losartán, EC50 >50 mM150,153,154. En realidad, el telmisartán parece ser el único ARB capaz de activar PPARg a las dosis terapéuticas usadas habitualmente150. Esto probablemente se debe a:
1.Su estructura química, pues el telmisartán es bastante distinto de los otros ARB aprobados para su uso clínico y comparte cierta homología estructural con la pioglitazona.
2.Su elevada lipofilia, que facilita su mayor difusión a través de las membranas celulares y hasta el compartimiento nuclear.
3.Su capacidad específica para interaccionar con residuos aminoacídicos específicos del LBD de PPARg153,154.
En resumen, los ARB, y particularmente el telmisartán, podrían ser útiles para el tratamiento del síndrome metabólico, debido a su efecto dual sobre la inhibición del receptor de angiotensina y la activación de PPARg. Estudios clínicos a pequeña escala y estudios retrospectivos han indicado que la administración de telmisartán mejora el metabolismo de la glucosa y de los lípidos, sin los efectos adversos provocados por los agonistas de PPARg totales. En pacientes con DM2 hipertensos, la administración diaria de 40-80 mg de telmisartán redujo la glucosa y los TG en plasma después del tratamiento160,161. No obstante, aún se requieren estudios adicionales a mayor escala, algunos de ellos ya en desarrollo, para comprender completamente el impacto de los ARB sobre el perfil metabólico de pacientes con elevado riesgo cardiovascular y también para elucidar la razón de las diferencias observadas in vitro entre los distintos ARB.
Antagonistas de PPARg
Aunque inicialmente se esperaría que los antagonistas de PPARg produjeran diabetes y lipodistrofia, podrían ser útiles para el tratamiento del síndrome metabólico debido a su potencial capacidad para inhibir la diferenciación adipocitaria. Como ya se ha descrito anteriormente, el SPPARM GW0072 puede bloquear los efectos inductores de la adipogénesis de la rosiglitazona sin alterar sus efectos sobre la insulina, demostrándose así una cierta actividad antagonista de PPARg145. El CDDO, o ácido 2-ciano-3,12-dioxoolean-1,9-dien-28-oico, es otro SPPARM que inhibe la diferenciación adipocitaria a bajas concentraciones (< 1 mM)162. El inhibidor de la ciclooxigenasa diclofenaco (10 mM) y el derivado plástico BADGE (100 mM) son antagonistas de PPARg que bloquean la adipogénesis mediada por rosiglitazona e insulina50,163. Recientemente se han descrito otros antagonistas de PPARg: LG100641 y SR-202. LG100641 incrementa la captación de glucosa e inhibe la diferenciación adipocitaria inducida por TZD en adipocitos 3T3-L1164, y SR-202 inhibe la activación de PPARg por parte de las TZD, así como el reclutamiento de SRC-1. SR-202 reduce la hipertrofia adipocitaria y la resistencia a la insulina inducidas mediante la dieta en ratones C57/BL6, y mejora la sensibilidad a insulina en ratones ob/ob134,165.
Agonistas de PPAR duales y panagonistas
Aunque las TZD suprimen la hiperglucemia en pacientes con DM2, no presentan efectos importantes sobre los parámetros lipídicos. Por el contrario, los fibratos, que son agonistas de PPARa, mejoran la hipertrigliceridemia y los bajos valores de colesterol HDL pero no mejoran la hiperglucemia. Los efectos beneficiosos de una activación conjunta de PPARa y PPARg ya se ha demostrado mediante la administración combinada de un agonista de PPARg, la rosiglitazona, con un agonista de PPARa como es el fenofibrato1. En este estudio, la activación de PPARg mejoró la sensibilidad a la insulina, mientras que la activación de PPARa incrementó el catabolismo lipídico, mejorando el perfil lipídico más allá del efecto observado a través de la activación de las 2 isoformas de PPAR por separado. En consecuencia, los agonistas PPARa/g duales podrían ser útiles para mejorar simultáneamente la hiperglucemia y el per fil lipídico patológicos en pacientes con DM2. Basándose en esta premisa, la modulación de PPAR mediante coagonistas o agonistas duales (PPARg/a y PPARg/d) y, potencialmente, pan-agonistas PPARa/g/d, debería, en teoría, mejorar los beneficios terapéuticos. Los agonistas PPAR dua les con actividad PPARg/d incluyen L-165461, L-783483 y L-796449. Pocos datos se tienen acerca de estos coagonistas PPARg/d, aunque en principio se esperaría que tuvieran efectos beneficiosos similares a los de los coagonistas PPARa/g.
Compuestos representativos con actividad agonista dual PPARg/a son O325H, LSN862 y los glitazars (ragaglitazar, muraglitazar y tesaglitazar). Todos los glitazars muestran mayor afinidad para PPARg que para PPARa, y su afinidad para PPARa es aún mayor que la de los fibratos. El ragaglitazar, cuando se administra a ratas ZDF, es capaz de reducir la glucosa en plasma, los TG y la insulina, en comparación con la rosiglitazona, y mejorar el aclarado de TG y de colesterol en comparación con el agonista de PPARa fenofibrato166. En este mismo modelo, tanto el ragaglitazar como la pioglitazona mejoraron la sensibilidad a la insulina y la hiperglucemia, y redujeron los lípidos plasmáticos al mismo nivel167. Por otro lado, el ragaglitazar y la pioglitazona también mejoraron la función de las células b, pero el primero fue más efectivo como terapia preventiva para la diabetes167. Ambos tratamientos causaron incremento de peso y de masa de tejido adiposo, así como mayor secreción de leptina, contradiciendo estudios previos que sugerían un efecto negativo del ragaglitazar sobre el peso corporal167. El mantenimiento de la respuesta secretora de insulina en ratas ZDF parecería el mecanismo más probable mediante el cual los glitazars mejoran la sensibilidad a la insulina. Estudios clínicos realizados con pacientes con DM2 o insulinorresistentes han demostrado que el muraglitazar, ya sea como monoterapia o en combinación con la metformina, mejora el control glucémico y estimula la sensibilidad a la insulina168,169, mientras que el tesaglitazar mejora la homeostasis de la glucosa y el perfil lipídico en ayuno170. Desafortunadamente, el ragaglitazar se ha retirado de estos estudios a consecuencia de sus peligrosos efectos adversos25,146. De la misma manera, se ha recomendado que el muraglitazar no se apruebe para el tratamiento de la diabetes hasta que no se realicen estudios de seguridad en ensayos donde se analicen los episodios cardiovasculares171.
O325H es un derivado del ácido propiónico con actividad agonista dual PPARg/a que se obtuvo por cribado computacional. Su administración en ratones db/db redujo los valores plasmáticos de TG, de colesterol total y de glucosa, sugiriendo que podría mejorar la sensibilidad a la insulina y reducir la hiperlipemia mediante la activación de ambos receptores nucleares, PPARg y PPARa172. LSN862, derivado del ácido 2-metoxidihidrocinámico, es un agonista dual PPARg/a de alta afinidad que también redujo la glucosa y los TG en modelos murinos de DM2 y dislipemia (ratas ZDF y ratones db/db)173. Asimismo, LSN862 incrementó la secreción de adiponectina en comparación con la rosiglitazona sin estimular el aumento de peso173. Puesto que LSN862 es un agonista parcial PPARg con menor, aunque significativa, actividad agonista de PPARa, podría ser un agente terapéutico muy útil para el tratamiento de la DM2 y su dislipemia asociada.
Conclusiones y perspectivas
El estudio de los efectos coordinados de las TZD sobre la activación de PPARg ha aumentado considerablemente el conocimiento de la función, de la selectividad y de la importancia biológica de este receptor nuclear. El descubrimiento y síntesis de nuevas clases de compuestos con actividad agonista sobre PPARg ha mejorado aún más nuestro conocimiento sobre su activación, reclutamiento selectivo de coactivadores y especificidad tisular. De hecho, PPARg tiene múltiples papeles fisiológicos en otros tantos tejidos: regula la diferenciación y la funcionalidad del tejido adiposo, promueve la sensibilidad a la insulina, estimula el flujo de colesterol y la captación de lípidos en macrófagos y presenta propiedades antiinflamatorias. La activación de PPARg conduce a la formación de adipocitos blancos más pequeños y sensibles a la insulina, así como a la diferenciación de adipocitos marrones, que regulan la homeostasis energética. Asimismo, PPARg activa la expresión de muchos genes implicados en la captación, el almacenamiento y la oxidación de ácidos grasos en adipocitos, lo que contribuye a la acción sensibilizadora a la insulina de los agonistas de PPARg. PPARg también regula la secreción de las adipocitocinas derivadas del tejido adiposo, como por ejemplo IL-6, TNF-a, leptina, resistina y adiponectina, que funcionan como reguladores hormonales de la sensibilidad a la insulina en otros tejidos. En consecuencia, la activación de PPARg presenta múltiples beneficios sobre el síndrome metabólico.
Las TZD son fármacos usados actualmente como sensibilizadores a la insulina para el tratamiento y la prevención de la resistencia a la insulina y DM2, pues estimulan el consumo de glucosa en el músculo esquelético y reducen la gluconeogénesis hepática. Además, las TZD no sólo mejoran la sensibilidad periférica a la insulina y el metabolismo lipídico, sino que también mejoran la función endotelial y reducen la aterogénesis y la fibrinólisis. No obstante, el amplio espectro de acción de PPARg dificulta su aplicación como herramienta terapéutica, debido al gran número de efectos colaterales que afloran debido a su estimulación. La modulación selectiva de PPARg debería solventar este problema, permitiendo la optimización de la especificidad génica y tisular. Florecientes áreas de interés en el desarrollo de nuevos fármacos incluyen los SPPARM. Los SPPARM podrían ser útiles para el tratamiento de la DM2 y otras enfermedades metabólicas como la obesidad, la arteriosclerosis y la hipertensión, pues mantienen el efecto reductor de glucosa y lípidos de las TZD sin promover la adipogénesis, incremento de peso corporal o edema. Los ARB, por ejemplo, actúan no sólo como antagonistas del receptor de angiotensina II, sino también como moduladores selectivos de PPARg cuando se administran en las dosis terapéuticas requeridas para el tratamiento de la hipertensión. Esta modulación parcial de la activación de PPARg por parte de los ARB comprende el reclutamiento selectivo de cofactores y un perfil de expresión génica distinto, con un modesto efecto sobre el patrón de adipogénesis. Otra aproximación terapéutica prometedora son los agonistas duales PPARg/a y los agonistas pan-PPAR, que permiten corregir la hiperglucemia y la dislipemia aterogénica asociadas con la diabetes y, por lo tanto, ampliar el espectro de beneficios terapéuticos de los agonistas PPARg puros. La caracterización molecular de estos compuestos abrirá nuevas perspectivas en el desarrollo de sustancias futuras que actúen sobre PPARg. De hecho, los SPPARM podrían erigirse en terapias útiles para el tratamiento de la DM2, mientras que los agonistas duales de PPARa/g podrían ser útiles para el tratamiento simultáneo de la hiperglucemia y la dislipemia diabéticas. Curiosamente, la reducción de la actividad PPARg mediante antagonistas también podría resultar beneficiosa para mejorar el perfil metabólico en sujetos enfermos, por medio de la promoción de la b-oxidación de ácidos grasos a través de la modulación de la secreción de adipocitocinas. No obstante, todavía se necesitan muchos esfuerzos para definir la fisiología, la farmacología y la funcionalidad de PPARg, con el fin de comprender y mejorar los beneficios y los efectos adversos de la modulación de la actividad de PPARg.
Tampoco se debe excluir el uso de otras dianas terapéuticas potenciales para la regulación de los efectos mediados por PPARg, como son PGC-1a o receptor retinoide X (RXR). PGC-1a es un cofactor específico que regula la termogénesis mediada por PPARg en el TAM, promueve la biogénesis y la respiración mitocondrial en el músculo esquelético, lo que sugiere un papel en el metabolismo oxidativo, y también participa en la gluconeogénesis hepática. En consecuencia, PGC-1a podría ser un objetivo terapéutico importante, especialmente como fármaco antiobesidad y antidiabético. Por otro lado, los agonistas selectivos de RXR, que activan el heterodímero PPARg/RXR, aunque no son estrictamente activadores de PPARg podrían ser útiles paraión adipocitaria y la sensibilidad a la insulina en ratones in vivo. En ambos casos, el amplio espectro de acciones fisiológicas y la ausencia de especificidad tisular dificultarían de nuevo su eficacia clínica.