




Ageing is the main risk factor for cardiovascular disease (CVD). The increased prevalence of CVD is partly due to the global increase in life expectancy. In this context, it is essential to identify the mechanisms by which ageing induces CVD, with the ultimate aim of reducing its incidence. Both atherosclerosis and heart failure significantly contribute to age-associated CVD morbidity and mortality. Hutchinson-Gilford progeria syndrome (HGPS) is a rare genetic disorder caused by the synthesis of progerin, which is noted for accelerated ageing and CVD. This mutant form of prelamin A induces generalised atherosclerosis, vascular calcification, and cardiac electrophysiological abnormalities, leading to premature ageing and death, mainly due to myocardial infarction and stroke. This review discusses the main vascular structural and functional abnormalities during physiological and premature ageing, as well as the mechanisms involved in the exacerbated CVD and accelerated ageing induced by the accumulation of progerin and prelamin A. Both proteins are expressed in non-HGPS individuals, and physiological ageing shares many features of progeria. Research into HGPS could therefore shed light on novel mechanisms involved in the physiological ageing of the cardiovascular system.
El envejecimiento es el principal factor de riesgo de enfermedad cardiovascular (ECV) y su prevalencia está aumentando progresivamente debido en gran parte al incremento de la esperanza de vida a nivel mundial. En este contexto, es fundamental establecer cuáles son los mecanismos por los que el envejecimiento promueve el desarrollo de ECV, con el objetivo de reducir su incidencia. La aterosclerosis y la insuficiencia cardiaca contribuyen de manera significativa a la morbimortalidad por ECV asociada a la edad. El síndrome de progeria de Hutchinson-Gilford (HGPS) se caracteriza por un envejecimiento prematuro que cursa también con ECV acelerada. Se trata de un trastorno genético raro causado por la expresión de progerina, una forma mutada de la prelamina A. La progerina induce aterosclerosis masiva y alteraciones electrofisiológicas en el corazón, promueve el envejecimiento y finalmente la muerte prematura a una edad media de 14,6 años, principalmente por infarto de miocardio o ictus cerebral. En esta revisión se discuten las principales alteraciones estructurales y funcionales que afectan al sistema vascular durante el envejecimiento fisiológico y prematuro, así como los mecanismos que subyacen a la aterosclerosis y al envejecimiento exagerados inducidos por la prelamina A y la progerina. Dado que ambas proteínas se expresan en individuos sin HGPS y muchas de las características del envejecimiento normal se presentan en la progeria, la investigación en el ámbito del HGPS podría contribuir a la identificación de nuevos mecanismos implicados en el envejecimiento cardiovascular fisiológico.
Cardiovascular disease (CVD) is intrinsically linked with ageing and accounts for more than 30% of all deaths worldwide.1,2 Although a wide range of diseases are associated with ageing, CVD represents the biggest burden for the elderly, their caregivers and health systems.3 The high prevalence of CVD can be explained thanks to advances in its treatment, which has increased life expectancy in developed countries and contributed to the progressive ageing of the population. Although this is a remarkable achievement from an individual perspective, the resulting demographic changes represent a challenge for social protection and healthcare systems around the world. For example, the population of the United States of America over 65 years of age is expected to increase from 12% in 2010 to 22% by 2040.4 In 2050, it is estimated that 19 countries will have an over-80s population in excess of 10%, many of whom will be afflicted with CVD or other age-related diseases and be unable to live independently.5 The financial cost of treating patients with CVD is enormous and is expected to increase significantly in the coming years. By way of example, the member states of the European Union collectively spend more than four billion Euros every day on medical care.6 In similar fashion, the direct medical cost of CVD is expected to triple in the United States between 2010 and 2030, while indirect costs are set to rise by 61% due to the loss of productivity associated with these diseases.4 That is why there is a pressing need to identify the mechanisms by which ageing leads to deterioration of the cardiovascular system, irrespective of other risk factors, many of which are modifiable. This knowledge is essential to be able to offer effective and sustainable medical care to a rapidly-ageing population.
Shared characteristics of physiological ageing and Hutchinson-Gilford progeria syndromeFour key factors that cause cumulative damage and are responsible for ageing in mammals have been identified: genomic instability, telomere shortening, epigenetic abnormalities and the loss of proteostasis.7 These processes induce so-called antagonistic mechanisms, which include altered sensitivity to nutrients, mitochondrial dysfunction and cellular senescence. These in turn foster the development of so-called integral mechanisms, which are the primary instigators of the ageing process and include stem cell depletion and impaired intercellular communication. These ageing mechanisms have either been identified either by comparative analysis between juvenile individuals or animals and those that have aged naturally, or by interventional studies which analyse how the specific impairment of genetic pathways or biochemical processes affect life expectancy during physiological ageing.
There is evidence to suggest that research into Hutchinson-Gilford progeria syndrome (HGPS) (OMIM code 176670) could aid our understanding of cellular and molecular mechanisms that are responsible for normal ageing and its associated CVD. Patients with HGPS exhibit premature ageing associated with accelerated atherosclerosis and vascular calcification, together with the development of cardiac electrical conduction defects. These abnormalities tend to cause the death of afflicted patients in the early years of adolescence (life expectancy 14.6 years), generally due to myocardial infarction or cerebral stroke. The disease is caused by the expression of progerin, an anomalous variant of prelamin A which arises due to a de novo mutation in the LMNA gene8,9 (see below). Other progeroid syndromes associated with mutations that elicit the loss of function of the ZMPSTE24 gene and an abnormal accumulation of prelamin A have been reported.10 It is striking that all the characteristic factors of normal ageing proposed by López-Otín et al.7 have been reported in progeria animal models, some of which have also been identified in patients with HGPS.7,11–14 Normal ageing in subjects without HGPS is characterised by low levels of expression of prelamin A and progerin in various cell types and tissues,11–13 including the cells of the adventitia, tunica media and atherosclerotic lesions in the coronary arteries.15 It has been reported that both oxidative stress and telomere shortening, phenomena that are believed to contribute to normal ageing,7 promote the expression of prelamin A and progerin in normal cells.16,17 This review summarises the primary structural and functional defects of the vascular system that arise as part of the ageing process, as well as the molecular and cellular mechanisms involved, based on our recent review of how ageing affects the cardiovascular system as a whole.18
Vascular changes as part of physiological ageingAutonomic nervous system abnormalitiesAgeing causes significant alterations to the autonomic nervous system's regulation of the cardiovascular system. These alterations include an increase in sympathetic activity due to raised levels of catecholamines in the blood, as well as decreased β-adrenergic sensitivity and a reduced arterial baroreceptor reflex.19–21 Although the specific causes of these alterations are as yet unknown, some authors have suggested that the decreases in β-adrenergic sensitivity and arterial baroreceptor activity may trigger the increase in sympathetic activity as a compensatory mechanism.22,23
HypertensionThe traditional premise that ageing causes hypertension continues to be the subject of intense debate.22 Results from longitudinal studies have demonstrated that the sympathetic activity associated with ageing increases blood pressure19; however, it is initial blood pressure values in youth that determine whether this increase will exceed the threshold to be considered hypertension.24 Sun et al. highlight the aetiological role of vascular rigidity in blood pressure elevation and propose that rigidity of the large arteries reduces baroreceptor activity, which is then compensated by an increase in sympathetic activity, which causes blood pressure to increase.23 In the short term, acute or beat-to-beat regulation of blood pressure is achieved primarily through baroreceptor reflexes. Since age is associated with a reduction in baroreceptor reflex activity, ageing has also been associated with increased blood pressure variation.25,26
Vascular rigidity, endothelial dysfunction and atherosclerosisArterial rigidity and endothelial dysfunction are two of the main age-related vascular alterations.1–3
Endothelial dysfunction involves alterations in the control of vessel tone and permeability, which promotes pro-inflammatory processes.3 The bioavailability of nitrous oxide, one of the principle cellular mediators released by the endothelium, is reduced, which increases endothelial permeability and inflammation, triggering a positive feedback process which aggravates the phenomenon in the long term.27 Endothelial dysfunction plays a fundamental role in both the onset and progression of atherosclerosis and forms a part of the degenerative process that affects the large arteries during ageing.2,22,28 Ageing is therefore also associated with vascular remodelling that increases the size of the lumen and thickens the tunica intima and tunica media.29 The increase in intima-media thickness is an early symptom of atherosclerosis22 and an independent predictor of future cardiovascular events.30 Intima thickening begins and is maintained due to the recruitment of blood leukocytes, a process triggered by the activation of adhesion molecules in dysfunctional endothelial cells (ECs).31 Leukocytes in the neointima induce a complex local immune response that promotes further leucocyte recruitment and induces the migration of vascular smooth muscle cells (VSMCs) from the tunica media towards the growing atherosclerotic lesion. The VSMCs activated in the neointima transform from a “contractile” phenotype to a “synthetic” phenotype, characterised by dedifferentiation, proliferation and abundant secretion of extracellular matrix components.32,33
There is some controversy surrounding whether atherosclerosis is the result of the accumulations of risk factors with ageing or, conversely, whether ageing itself promotes atherosclerosis independently of other factors. The absence of atherosclerosis among the elderly of isolated tribal societies34 and its presence in children with a high degree of exposure35 demonstrates the significant influence of exposure to risk factors and indicates that it is possible to age without atherosclerosis. Nevertheless, signs of atherosclerosis have been detected in the remains of human mummies from societies not exposed to modern risk factors.36 This fact, together with the incidence of atherosclerosis in individuals with HGPS and other premature ageing syndromes, support the hypothesis that the most significant risk factor for atherosclerosis is ageing itself.
Arterial rigidity is another pathological characteristic of ageing that is considered to promote the onset and progression of hypertension and atherosclerosis by inducing dysfunction of ECs and VSMCs in the tunica media.37 Vascular rigidity can be measured non-invasively using pulse wave velocity. This parameter is a very reliable indicator of cardiac events and is independent of blood pressure in a range of adult populations, including the elderly.38 Carotid-femoral pulse wave velocity gradually increases from 50 years of age and may reach an incidence of 64% in men and 74% in women over 70 years of age.39,40 The increased rigidity of the large arteries increases cardiac effort, leading to fibrosis and heart failure.
The mechanisms that contribute to the increase in arterial rigidity during ageing include alterations to the extracellular matrix and an associated increase in fibrosis and inflammation.23 Modification of the structure and composition of the extracellular matrix of the vessel wall in response to ageing is due to the increase in the deposition and maturation of collagen, the accumulation of advanced glycation end products and the rupture of elastic fibres.20,37,41
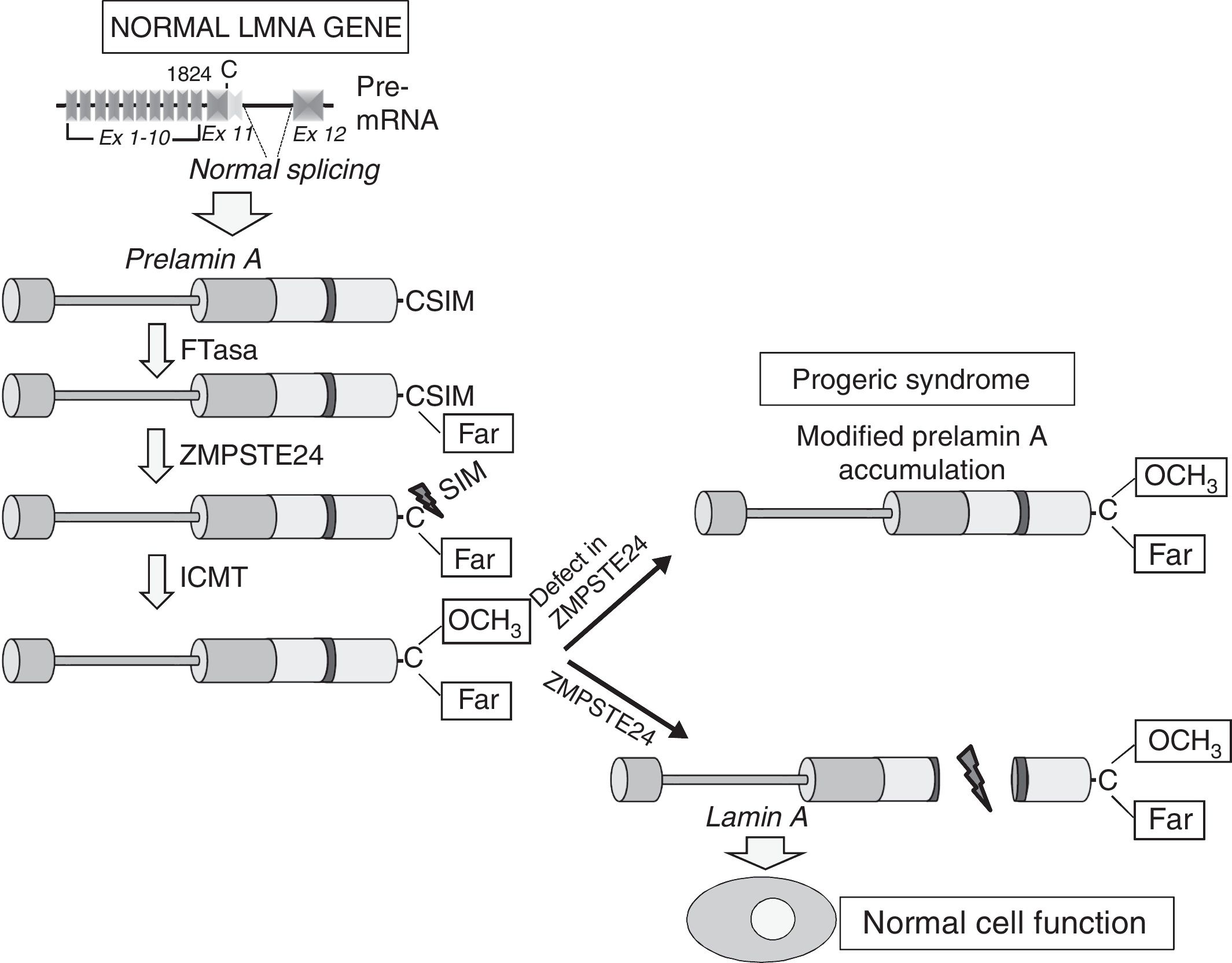
Cardiovascular disease in patients with Hutchinson-Gilford progeria syndromeHGPS is an ultra-rare human genetic disease with an estimated prevalence of one in every 20 million people (www.progeriaresearch.org). The disease is characterised by accelerated ageing caused by a de novo mutation in the LMNA gene.8,9 In normal cells, alternative splicing of the primary mRNA transcript of the LMNA gene gives rise to two main varieties of type A lamin (lamin A and lamin C), as well as less abundant varieties (lamin AΔ10 and lamin C2, specific to germ cells).42–44 The precursor proteins to lamin A, known as prelamin A, undergoes a number of post-translational modifications to give rise to the mature protein (Fig. 1). First, farnesyltransferase farnesylates cysteine residues in the cysteine–serine–isoleucine–methionine C-terminal domain. The three amino acids in the C-terminal position are then eliminated, which allows methylation of the new C-terminus catalysed by isoprenylcysteine carboxyl methyltransferase (ICMT). Finally, zinc-dependent metalloprotease ZMPSTE24/FACE-1 eliminates the 15 C-terminal residues together with the farnesyl and carboxymethyl group. The mature lamin A is then incorporated into the nuclear lamina, a network of structural proteins that is intimately associated with the inner nuclear membrane. In addition to providing mechanical resistance to the nucleus, type A lamins regulate multiple cell functions, including DNA replication and repair, chromatin organisation, signal transduction and gene transcription.45

Synthesis and processing of prelamin A in normal cells. Normal splicing between exons 11 and 12 of the LMNA gene enables synthesis of prelamin A, which undergoes a series of post-translational modifications culminating in the production of mature lamin A. Proteolytic prelamin A processing catalysed by the protease ZMPSTE24 eliminates its farnesylated and carboxymethylated C-terminus. Mutations that inactivate ZMPSTE24 lead to the accumulation of permanently farnesylated and carboxymethylated prelamin A and accelerate cellular ageing. Ex: exon; Far: farnesylated residue; FTase: farnesyltransferase; ICMT: isoprenylcysteine carboxyl methyltransferase.
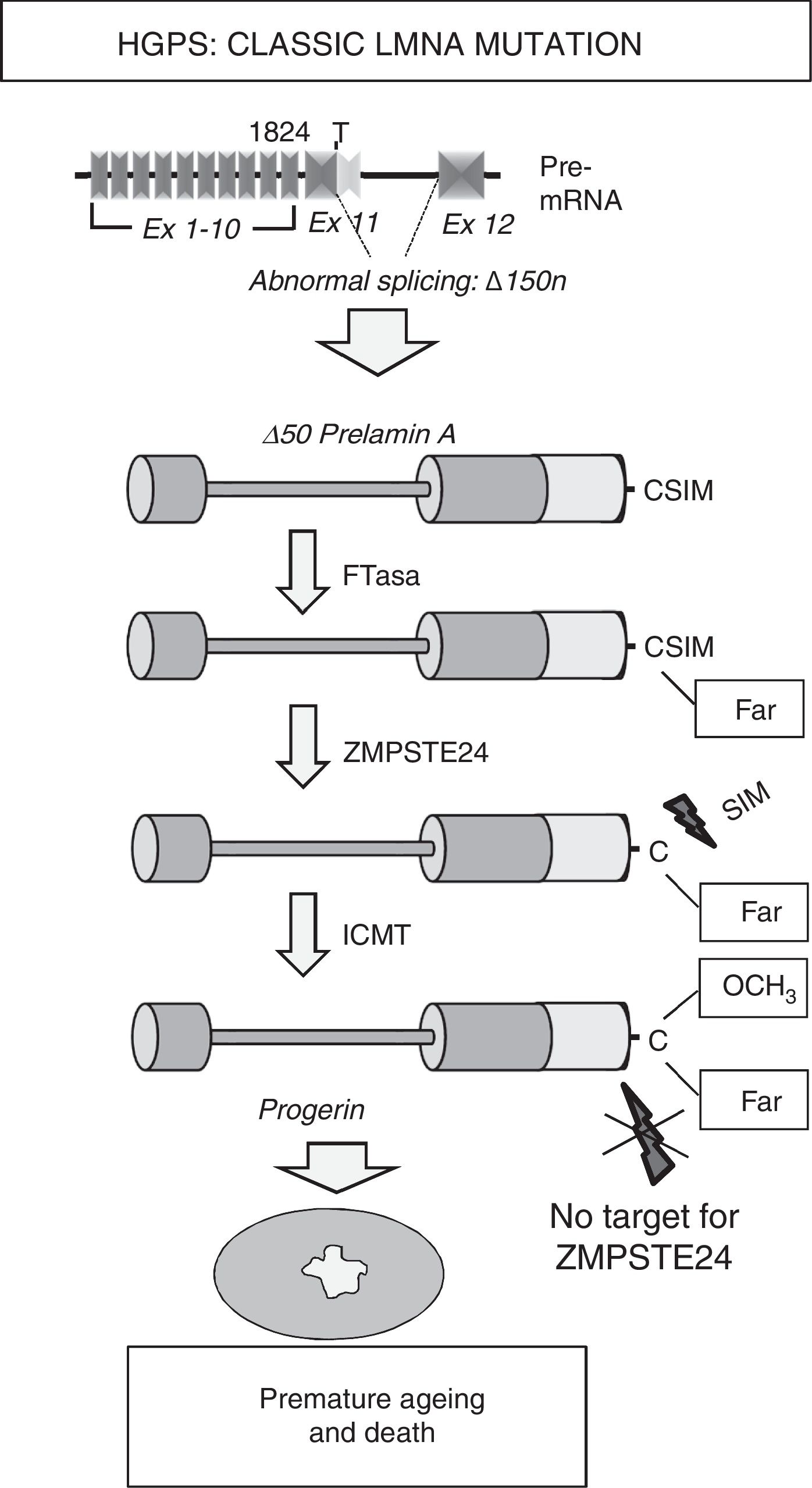
Most HGPS patients present the de novo heterozygotic point mutation c.1824C>T (p.G608G) in the LMNA gene.8,9 Although this mutation, responsible for “classic” progeria, is synonymous, it creates an aberrant splicing site on exon 11 that eliminates 150 nucleotides, giving rise to the synthesis of a truncated variant of prelamin A known as progerin (Δ50 prelamin A) (Fig. 2). The missing 50 amino acids at progerin's C-terminus prevents the excision of the 15 terminal amino acids, meaning that patients’ cells accumulates permanently farnesylated and carboxymethylated progerin. Similarly, mutations that inactivate the ZMPSTE24/FACE-1 gene also provoke the accumulation of farnesylated, carboxymethylated prelamin A and are associated with the development of progeroid syndromes in humans.10
 that leads to abnormal splicing and progerin synthesis. The absence of the region of 50 amino acids that includes the recognition site for ZMPSTE24 prevents the elimination of progerin")
Synthesis and processing of prelamin A in HGPS patients. Classic progeria is associated with a de novo heterozygotic point mutation in the LMNA gene (c.1824C>T) that leads to abnormal splicing and progerin synthesis. The absence of the region of 50 amino acids that includes the recognition site for ZMPSTE24 prevents the elimination of progerin's C-terminus, which remains permanently farnesylated, causing multiple cellular alterations that lead to premature ageing and death. Ex: exon; Far: farnesylated residue; FTase: farnesyltransferase; ICMT: isoprenylcysteine carboxyl methyltransferase.
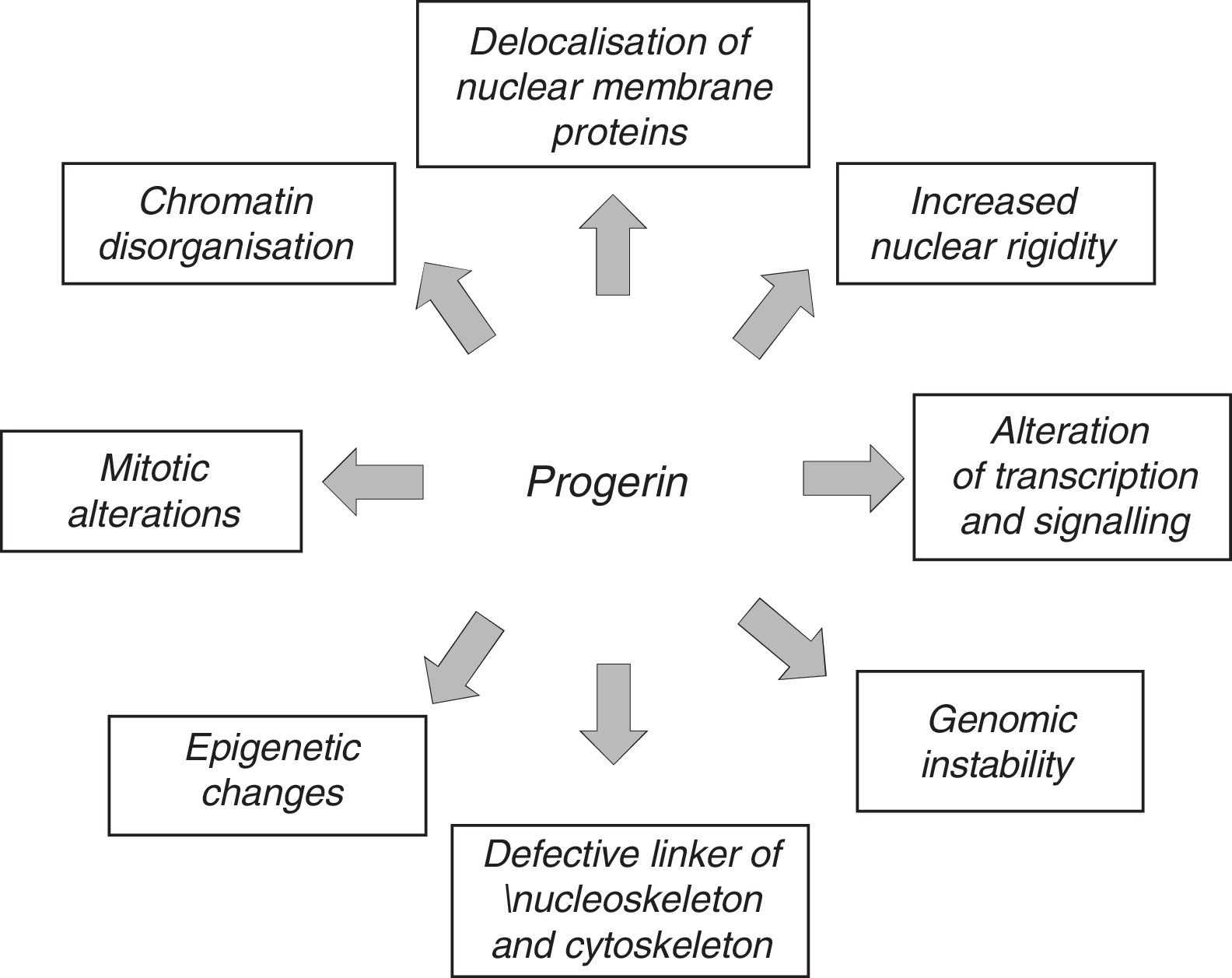
The anomalous expression of prelamin A and progerin causes multiple functional and structural alterations that affect signal transduction, gene transcription, chromatin organisation and, finally, slow down cell proliferation, causing senescence and cell death and accelerating the ageing of the body (Fig. 3).12,45 It should be noted that many of the processes involved in HGPS also play a role in normal ageing.7

Patients with HGPS have a normal appearance at birth but begin to develop symptoms during the first 12–18 months of life. The disease is characterised by stunted growth, abnormal dentition, alopecia, lipodystrophy, skin anomalies, joint contractures, osteoporosis and osteolysis that gradually lead to difficulties walking and performing other motor activities. Nevertheless, the most serious medical problems in patients with HGPS are atherosclerosis and cardiac electrical conduction defects, which lead to premature death at a mean age of 14.6 years, primarily due to myocardial infarction or stroke.46–50
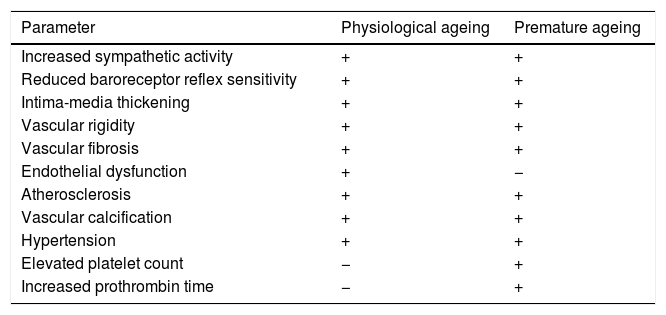
Many of the cardiovascular alterations in HGPS patients are also found in physiological ageing (Table 1). However, unlike the elderly, HGPS patients present an elevated platelet count and prothrombin time.49 The physiological causes and effects of these alterations are still unknown, and there is therefore a need for more specific studies that could determine their possible relevance. Moreover, HGPS patients often do not have traditional cardiovascular risk factors or are only partially affected by them. For example, HGPS patients have plasma cholesterol, LDL and HDL cholesterol, triglyceride and C-reactive protein levels similar to healthy children.48,49,51 In addition, some 30% of HGPS patients only present slight increases in systolic and diastolic blood pressure compared to healthy children of the same age.48,49,52 The study of HGPS-associated CVD therefore offers a unique opportunity to identify the mechanisms that cause age-related cardiovascular damage in the absence of other risk factors or other chronic diseases associated with ageing that might have a secondary impact on cardiovascular health.
Structural and functional changes in the vascular system during physiological and premature ageing.
| Parameter | Physiological ageing | Premature ageing |
|---|---|---|
| Increased sympathetic activity | + | + |
| Reduced baroreceptor reflex sensitivity | + | + |
| Intima-media thickening | + | + |
| Vascular rigidity | + | + |
| Vascular fibrosis | + | + |
| Endothelial dysfunction | + | − |
| Atherosclerosis | + | + |
| Vascular calcification | + | + |
| Hypertension | + | + |
| Elevated platelet count | − | + |
| Increased prothrombin time | − | + |
+: presence; −: absence.
As with normal ageing, non-invasive imaging techniques in HGPS patients can only detect evident carotid plaques in advanced stages.48,49,52 However, arterial stenosis affects HGPS patients of all ages and it has been proposed that it might be an early indicator of plaque formation.48 Atherosclerotic plaque in HGPS patients is accompanied by other vascular alterations that are typical of normal ageing, such as inflammation, loss of VSMCs and fibrosis in the tunica media, and the erosion and rupture of plaque.15 Conversely, one characteristic of the vessels of HGPS patients that is not common in those of control subjects is the presence of significant thickening of the adventitia and fibrosis.15,53 Another characteristic of physiological ageing that is also found in HGPS patients is vessel calcification, which is associated with an increase in morbidity/mortality due to CVD in the general population.54 Calcification affects the aorta and aortic and mitral valves of some HGPS patients and can lead to aortic or mitral regurgitation.15,49,55–58 Neuroimaging studies in a cohort of 25 patient detected the presence of early and clinically silent strokes, suggesting that this is a prevalent characteristic in HGPS.59 However, stroke in HGPS patients can also cause neurological sequelae.60
HGPS is also characterised by vascular rigidity, a change that is associated with physiological ageing and that predicts the incidence of future cardiovascular events independently of other factors.38 The analysis of a cohort of 21 patient with HGPS determined that vascular rigidity is an early and generalised phenomenon in this disease: the pulse wave velocities detected were comparable to those usually found in adults over 60 years of age.48 Even if intima-media thickness in the carotid artery of HGPS patients is normal, its echodensity is higher than normal, especially in the adventitia, in keeping with the vascular fibrosis detected in autopsies.15,48,49,61 HGPS patients present alterations in the ankle-brachial pressure index, an indicator of peripheral artery disease and vascular dysfunction48,56; however, non-invasive assessment of endothelial function by measuring flow-mediated vasodilation appears to indicate that patients do not present endothelial dysfunction.49 This data highlights the need to conduct additional studies analysing in detail the mechanisms that trigger progerin-induced vascular dysfunction.
In summary, the main characteristics of CVD in HGPS patients are vascular rigidity and remodelling, with significant fibrosis of the tunica media and adventitia, loss of VSMCs, accelerated atherosclerosis and premature death due to myocardial infarction or stroke. Premature death in HGPS may also be associated with arrhythmias deriving from cardiac electrical conduction defects, an aspect that is outside the scope of this review.12,18 New studies are required in order to precisely define the mechanisms through which progerin expression accelerates CVD and to clarify the relative contribution of vascular and cardiac alterations to premature death in HGPS.
Mechanisms and treatment of Hutchinson-Gilford progeria syndromeLamins A and C play a fundamental role in a wide range of cell functions, including maintenance of the mechanical stability of the nucleus, signal transduction, gene transcription, chromatin organisation, DNA repair, cell cycle progression and cell differential and migration.45 The accumulation of progerin and prelamin A therefore accelerates ageing through a number of mechanisms that affect multiple pathways and are not mutually exclusive. These mechanisms activated by prelamin A and progerin may differ between distinct tissues depending on differences in the expression of lamin A (and therefore of prelamin A and progerin), which is related to tissue rigidity.62
Studies in humans and mice suggest that the severity of HGPS is determined by both the total quantity of progerin and by the proportion of farnesylated progerin versus mature lamin A. In fact, different human LMNA point mutations are associated with significant differences in progerin level and disease severity, ranging from neonatal progeria (high progerin levels) to late-onset progeria (low progerin levels).63–65 Likewise, in LmnaG609G/G609G mice who carry a mutation in their endogenous Lmna gene that is equivalent to the c.1824C>T (p.G608G) mutation that causes HGPS in humans, an improvement in the ageing phenotype and increased survival were observed when progerin expression was reduced by using morpholino antisense oligonucleotides to block anomalous splicing between exons 11 and 12.66 In an analogous fashion, treatment with antisense oligonucleotides designed to promote alternative splicing of lamin A to lamin C also reduced progerin production, fibrosis of the adventitia and loss of VSMCs in LmnaG609G/G609G mice, although no effect on longevity was described for this strategy.67
Unlike mature lamin A, progerin remains permanently farnesylated (Fig. 1). The hypothesis that persistent farnesylation is the principle cause of progeria is based on studies in mice and humans that have demonstrated a positive association between progeroid symptoms and the accumulation of farnesylated prelamin A as a result of ZMPSTE24 deficiency.68–71 Similarly, a patient who had both a homozygotic mutation resulting in loss of function in ZMPSTE24 and a heterozygotic mutation in the LMNA gene giving rise to elongation of the C-terminus of lamin A, presented a milder than usual progeroid phenotype, possibly due to reduced levels of farnesylated prelamin A.72 The fact that Zmpste24−/− mice with LMNA haploinsufficiency do not present an evident ageing phenotype supports this conclusion.73 The importance of farnesylation in the pathogenesis of HGPS has been confirmed thanks to the generation of LmnacsmHG/csmHG mice, which produce non-farnesylated progerin and do not die prematurely.74 Likewise, LmnanPLAO/nPLAO mice who express only non-farnesylated prelamin A develop cardiomyopathy but no progeria.75 Treatment with farnesyltransferase inhibitors also reduces the nuclear alterations observed in cells that express progerin76,77 and prevents the onset and later progression of CVD in G608G BAC progeroid mice,78 which contain the human version of the LMNA gene with the c.1824C>T (p.G608G) mutation that causes HGPS.79 In an analogous fashion, these inhibitors prolong the survival of Zmpste24−/− and LmnaHG/+ progeroid mice, characterised by expressing prelamin A and progerin respectively.79,80
Subsequent studies showed that treatment with a farnesyltransferase inhibitor led to alternative prenylation of prelamin A and progerin mediated by geranylgeranyltransferase.81 Combined treatment with statins and aminobisphosphonates to block both farnesylation and geranylgeranylation of prelamin A improved the progeroid phenotype and prolonged the life of Zmpste24−/− mice.82 Based on the findings in cells and mice that express progerin and prelamin A,11 clinical trials have been conducted in patients with HGPS to analyse the effect of treatment with a farnesyltransferase inhibitor (lonafarnib) alone or combined with statins (pravastatin) and bisphosphonates (zoledronate).46,52,61 Lonafarnib monotherapy resulted in some improvement of vascular rigidity, bone structure and auditory status, and it was estimated that it increased survival by 1.6 years.46,61 Treatment with all three drugs in combination (lonafarnib, pravastatin and zoledronate: triple drug trial) resulted in an additional improvement in bone density but there was no improvement in cardiovascular parameters in comparison with lonafarnib monotherapy.52 Therefore, although farnesylated progerin appears to play a fundamental role in HGPS, current therapies that seek to prevent progerin farnesylation only provide moderate benefits.
The observation that both farnesylated and non-farnesylated progerin accumulate in the nuclear membrane pushed Kalinowski et al. to propose that the association of progerin with the inner nuclear membrane also involves increases in electrostatic interactions and aggregation.83 Moreover, the properties of the progerin tail, which is less heterogeneous and more compact that normal lamina A, may alter its interaction with DNA and other proteins.84
Another factor that might contribute to progerin's toxicity is the alteration of its protein structure due to the elimination of 50 amino acids near the C-terminal region. Progerin's abnormal interactions with other nuclear components lead to nuclear blebbing, increased thickness and rigidity, anomalous localisation of heterochromatin, and the alteration of nuclear pore complexes.85,86 Recently, Lee et al.87 described how progerin interacts strongly with lamin A/C and how the chemical disruption of progerin-lamin A/C heterodimers reduces nuclear aberrations, prevents cellular senescence, limits progeroid traits and lengthens life expectancy in LmnaG609G/G609G mice.
Carboxymethylation of progerin's C-terminal farnesylcysteine catalysed by ICMT may also contribute to progeria. The 70–90% reduction in ICMT expression and activity in hypomorphic Zmpste24−/−lcmthm/hm mice improved body weight, grip strength and bone structure and lengthened survival in comparison with Zmpste24−/−lcmt+/+ control mice with intact ICMT.88 The decrease in ICMT activity in Zmpste24−/−lcmthm/hm mice was associated with an alteration in the localisation of prelamin A and with the activation of AKT-mediated signalling (protein kinase B) and mTOR (mammalian target of rapamycin), which in turn delays cellular senescence. Nevertheless, it should be noted that rapamycin, an mTOR inhibitor, activated progerin elimination through autophagia and reduced nuclear abnormalities.89,90 These results clearly show that ICMT and mTOR are involved in premature ageing, although new studies will be required in order to precisely define the mechanisms involved and the relationship between ICMT, AKT and mTOR.
Mechanisms involved in the development of cardiovascular disease in progeriaThis section brings together the information available to date on the cellular and molecular mechanisms through which prelamin A and progerin damage the cardiovascular system. This knowledge is fundamental to our understanding of the mechanisms involved in the development of CVD during normal ageing, since both prelamin A and progerin are expressed at low levels in the cells and tissues of healthy individuals without progeria, including VSMCs in the tunica media and atherosclerotic lesions.15,16,91
Loss of vascular smooth muscle cellsGradual loss of VSMCs is a characteristic of HGPS patients15,53,92 and progeria models in mice,66,67,79 which points to its contribution in progeroid vascular disease. Although less serious, reduced VSMC content in the tunica media is also seen in physiological ageing.93
VSMCs are subjected to elevated mechanical stress associated with blood flow. Under normal conditions, cells respond to increased shear stress by increasing lamin A/C expression and modifying the location of their nucleus.62,94,95 In cells that express progerin, abnormal responses to physical stress may lead to damage and cell death.86,96 In line with this idea, it has been described how mechanical stress applied to fibroblasts of HGPS patients reduces viability and increases cell death by apoptosis.97 Progerin-induced alterations to mechanotransduction could be explained by changes in the expression of proteins that control cytoskeletal organisation, mechanotransduction and production of the extracellular matrix.98,99 In fact, the ascending aorta of progerin-expressing BAC G608G transgenic mice presents a reduction in the expression of vimentin, a cytoskeletal protein that binds to the nucleus, endoplasmic reticulum and mitochondria, and is essential for maintaining the integrity of the cell.100 This correlation between reduced expression of mechanotransduction proteins and increased shear stress may partially explain the loss of VSMCs in HGPS.
The mechanisms underlying the loss of VSMCs caused by progerin can be analysed using differentiation protocols on induced pluripotent stem cells (iPSCs) from healthy individuals and HGPS patients. Liu et al. observed that VSMCs differentiated from iPSCs that expressed progerin presented premature senescence as a consequence of the interaction of progerin with the catalytic subunit of DNA-dependent protein kinase, a catalytic subunit of nuclear DNA-PK which plays a role in DNA repair through non-homologous recombination.101 Conversely, Kinoshita et al. described that progerin, unlike lamin A, cannot interact with DNA-PK or other proteins involved in the response to DNA damage.102 They also observed that the expression of progerin in VSMCs, but not in ECs, induces DNA-PK activation, which slows the growth of VSMCs and induces senescence. In view of these discrepancies, it is evident that new studies are needed to explain the interaction between progerin and DNA-PK and its subunits in different types of cells and to establish the pathophysiological consequences.
Zhang et al. described a defective proliferation of VSMCs derived from iPSCs from HGPS patients as a consequence of independent caspase activation mechanisms.103 These authors also observed that progerin expression in VSMCs inhibits poly(ADP-ribose)-polymerase 1, a key regulator of DNA repair, and that it activates the error-prone, non-homologous recombination response, which prolongs mitosis and provokes mitotic catastrophe and cell death. Prelamin A also induces DNA damage and increases the cell response to DNA damage in aged VSMCs.16,104 This response may be the result of defective recruitment of p53 binding protein 1 (53BP1) to damaged regions of DNA, which disrupts transport to the nucleus due to an alteration in the localisation105 of nucleoporin 153. Alteration of the repair of damaged DNA has also been described in non-vascular cells from HGPS patients and in progeria models in mice.106–108 All of these findings confirm that progerin-induced VSMC death is at least partly due to defects in DNA repair, an alteration that also plays a fundamental role in normal ageing.109
Vascular calcificationAs in HGPS patients,15,55–57G608G BAC and LmnaG609G/+ progeroid mice develop aortic calcification.79,110 Calcified aortas in LmnaG609G/+ mice show abnormally elevated expression of osteogenic markers, including bone morphogenic protein 2 (Bmp2) and Runt-related transcription factor 2 (Runx2), without any alteration being present in anticalcifying agents such as Matrix Gla protein and fetuin A.110 Similarly, primary VSMCs derived from LmnaG609G/+ mice showed a lower capacity to inhibit calcium deposition in vitro, which was associated with a lower concentration of extracellular inorganic pyrophosphate (ePPi), the main endogenous inhibitor of vascular calcification. The reduction in ePPi levels in VSMC cultures was associated with a reduction in their synthesis caused by decreased production of ATP (the main substrate in ePPi synthesis) and the induction of both tissue-nonspecific alkaline phosphatase (the fundamental enzyme in PPi hydrolysis) and ectonucleoside triphosphatase diphosphohydrolase 1 (an enzyme that hydrolyses ATP to release PPi). In fact, the LmnaG609G/+ mice showed plasma concentrations of ePPi and ATP below those of their Lmna+/+ litter-mates, while treatment with exogenous PPi prevented vascular calcification in the LmnaG609G/G609G mice.110
Expression of prelamin A in VSMCs promotes vascular calcification by activating the ataxia-telangiectasia-mutated/ataxia-telangiectasia and Rad3-related signalling pathway,104 which is associated with DNA damage. The activation of this pathway in VSMCs induces the secretor phenotype associated with cellular senescence, releasing procalcifying factors such as BMP2 which can trigger calcification both locally and in remote regions.104 Furthermore, VSMC cultivation in a procalcifying medium entails the induction of lamin A and prelamin A expression, together with an increase in the expression of procalcifying factors such as Runx2, osteocalcin and osteopontin and an increase in calcium deposition.111 It should be noted that human mesenchymal stem cells that express progerin also present elevated levels of osteopontin and show an increase in osteogenic differentiation.112
In summary, lamin A and its mutated or unprocessed forms play a role in osteoblast differentiation and in the calcification of VSMCs, which demonstrates the need to deepen our knowledge surrounding the role of progerin and prelamin A in vascular calcification in premature ageing.
Endothelial dysfunctionEndothelial dysfunction plays a fundamental role in all phases of atherosclerosis, the disease that compromises the lives of HGPS patients. ECs detect and respond to different types of blood flow, and the aortic regions that are subject to a turbulent blood flow and elevated shear stress, such as the ascending aorta, are more susceptible to developing atherosclerosis.113,114 Song et al. detected intact EC monolayers in ascending zones of the aorta in G608G BAC mice with almost no VSMCs.98 Progerin-expressing ECs close to regions that have suffered a massive loss of VSMCs present vimentin expression that is 8 times higher than that of ECs located in regions with preserved VSMC content, which makes them more resistant to shear stress and may explain the presence of well-preserved endothelium in the vessels of HGPS patients.15
The elevated expression of adhesion molecules in dysfunctional ECs results in the adhesions of monocytes, an important stage in the onset and progression of atherosclerosis.31 Recent studies show that accumulation of prelamin A in ECs due to blocking of lamin A maturation induces cellular senescence and promotes monocyte adhesion based on intercellular adhesion molecule 1.115 It is evident that characterising the connection between ECs, progerin and prelamin A expression and the progression of atheromatous plaque will require new studies.
Final conclusions and perspectivesAgeing is the principal risk factor for CVD. Faced with the progressive ageing of our society, and given that CVD constitutes the principal cause of morbidity and mortality globally, we urgently need to improve our knowledge of the mechanisms that contribute to ageing. This information is fundamental to the development of new strategies to reduce the onset and progression of disease in old age, and therefore to foster healthy ageing. The intensive work performed in the fields of fundamental, clinical and epidemiological research has enabled us to identify the general mechanisms involved in ageing. The challenge in research on ageing is to identify which of these mechanisms contribute primarily to explaining the high inter-individual variability in human biological ageing. This knowledge will contribute to the development of new therapies and to improving prevention through the early identification of those individuals at greatest risk of suffering from age-related diseases, before the first symptoms appear. These actions will foster healthy ageing and reduce the socioeconomic impact on health systems caused by ageing.
Ageing and CVD are highly accelerated in patients with HGPS, a very rare genetic disorder caused by progerin, an unprocessed form of lamin A. Progerin in humans is also associated with the abnormal accumulation of prelamin A due to mutations that inactivate the protease ZMPSTE24. It should be noted that both prelamin A and progerin are expressed, albeit at low levels, in the cells and tissues of elderly individuals, including the cells of vessel walls. In the cells of individuals without HGPS, it has been proposed that oxidative stress and telomere shortening induce the expression of prelamin A and progerin, respectively. Research into progeria may therefore help us to identify the cellular and molecular mechanisms that govern normal ageing and associated CVD. Traditional cardiovascular risk factors such as hypercholesterolaemia, diabetes, obesity, hypertension and smoking are often absent or mild in HGPS patients. Research into this disease therefore offers a unique opportunity to differentiate age-dependent mechanisms that directly induce cardiovascular damage from modifiable risk factors that cause gradual deterioration of cells and tissues during old age and can have a secondary impact on cardiovascular health.
Identification of both the specific and common mechanisms involved in the progression of normal and premature ageing will require large-scale genomics, epigenomics, transcriptomics, proteomics and metabolomics studies. It will also be possible to establish causal relationships through strategies that use functional losses and gains targeting the candidate factors identified in the “omics” studies. Considering the large number of cell types that play a role in CVD and in normal and premature ageing, the generation of new conditional or tissue-specific animal models, with particular emphasis on cells that play a critical role in atherosclerosis (e.g. monocytes/macrophages, lymphocytes, ECs and VSMCs). Because the animal progeria models generated to date do not develop atherosclerosis, the primary cause of death in HGPS patients, the generation of progeria models, in both small and large animals, that do present atherosclerosis will be essential. Future challenges will focus on establishing how physiological ageing leads to the accumulation of unprocessed forms of lamin A and on determining whether the nuclear anomalies induced by these proteins contribute to normal ageing.
FundingThe laboratory work carried out by V.A. was funded by the Ministry of Economy, Industry and Competition (MEIC; SAF2016-79490-R) and the Instituto de Salud Carlos III (ISCIII, Spanish public health research institute) (RD12/0042/0028 and AC16/00091), with co-financing from the European Regional Development Fund (ERDF), TV3's Fundació Marató (122/C/2015) and the Progeria research Foundation (Established Investigator Award 2014-52). The Biomedical Research Network Centres (CIBER) for Cardiovascular Diseases is an ISCIII initiative.
L.C. received assistance from the ISCIII Cardiovascular Research Network's Jordi Soler postdocotral grants programme, and M.R.H. received predoctoral assistance from the MEIC's FPI (research staff training) programme (BES-2011-043938). The CNIC (Spanish national cardiovascular research centre) receives support from the MEIC and the Fundación Pro-CNIC and has been accredited as a “Severo Ochoa” Centre of Excellence (MEIC SEV-2015-0505).
The authors apologise to all those researchers whose work could not be cited due to length limitations.
Please cite this article as: del Campo L, Hamczyk MR, Andrés V, Martínez-González J, Rodríguez C, en nombre del Grupo de trabajo de Biología Vascular de la Sociedad Española de Arteriosclerosis. Mecanismos de envejecimiento vascular: ¿Qué podemos aprender del síndrome de progeria de Hutchinson-Gilford? Clin Investig Arterioscler. 2018;30:120–132.