




El arco aórtico derecho es una entidad rara (prevalencia del 0,1%) que se adscribe a un grupo de malformaciones poco frecuentes y escasamente descritas en la bibliografía en su forma prenatal; sin embargo, no es infrecuente hallarlas como causa de enfermedad respiratoria o digestiva (refractarias a tratamiento) en pacientes adultos, e incluso como procesos vasculares severos con morbimortalidad elevada. Sus variantes pueden condicionar sintomatología compresiva y estar relacionadas con anomalías cardíacas (hasta 90%) y/o con la microdeleción 22q11 (hasta un 46%). El corte ecográfico de 3 vasos-tráquea es fundamental para su diagnóstico prenatal, y tras su detección está indicada una evaluación exhaustiva del corazón fetal, distinguir sus variantes y/o un estudio cromosómico-genético específico. En presencia de otras anomalías el pronóstico es pobre; los casos aislados suelen tener una evolución posnatal oligo o asintomática.
Right aortic arch, which belongs to a group of rare malformations, is an uncommon finding (incidence of 0.1%) and few cases of prenatal forms have been described in the literature. Nevertheless, it is not unusual to find these anomalies as the cause of respiratory or digestive disease (refractory to treatment) in adult patients and even as severe vascular processes with high morbidity and mortality. Its variants may cause compressive symptoms and be associated with heart defects (up to 90%) and/or 22q11 microdeletion (up to 46%). The 3 vessels and trachea view is essential for the prenatal diagnosis of right aortic arch. Detection of this condition must be considered an indication for foetal echocardiography, to distinguish its variants, and/or specific chromosome/genetic testing. Isolated right aortic arch is usually asymptomatic, although the outcome of right aortic arch associated with other abnormalities is poor.
El diagnóstico ecográfico de las cardiopatías congénitas (CC) sigue siendo uno de los retos del diagnóstico prenatal. A pesar de los avances médicos, que han permitido un mejor conocimiento anatómico-ecográfico del corazón fetal, y a los avances tecnológicos, que han mejorado la resolución de los ecógrafos, las CC en buena parte siguen pasando inadvertidas durante la vida fetal1–3. El conocimiento anatómico y funcional del corazón fetal ha aumentado desde la aparición e implantación de la ecografía como método de diagnóstico prenatal. Así, desde la descripción de la imagen de las 4 cámaras cardíacas hasta los 5 planos de Yagel4, la ecografía ha permitido una mejora ostensible en el diagnóstico de las CC y puede detectar malformaciones que hasta hace poco pasaban inadvertidas. Durante el desarrollo embrionario, el sistema aórtico con 2 vasos simétricos, uno izquierdo y otro derecho, forma un anillo vascular rodeando el tubo digestivo-pulmonar en crecimiento. Habitualmente, el arco aórtico derecho (AAD) presenta una regresión y el izquierdo prevalece1 (manuscrito anónimo). Las alteraciones del desarrollo del arco aórtico se presentan en el 1-2% de la población general, donde el 10% son AAD2,3,5 y pueden presentarse aisladas o asociadas con anomalías cardíacas, extracardíacas y/o cromosomopatías (especialmente microdeleciones en el cromosoma 22q11 [CATCH-22])1,6. El diagnóstico ecográfico prenatal del AAD se realiza mediante un corte transversal del tórax fetal a nivel mediastínico, cuando el arco aórtico se sitúa a la derecha de la tráquea5. Se presentan 2 casos de AAD, que se detectaron al aplicar como método de cribado para el estudio cardíaco la visualización de los 5 planos de Yagel durante la práctica de la ecografía morfológica, método empleado en todas las embarazadas controladas en nuestro servicio en torno a la semana 20 de embarazo.
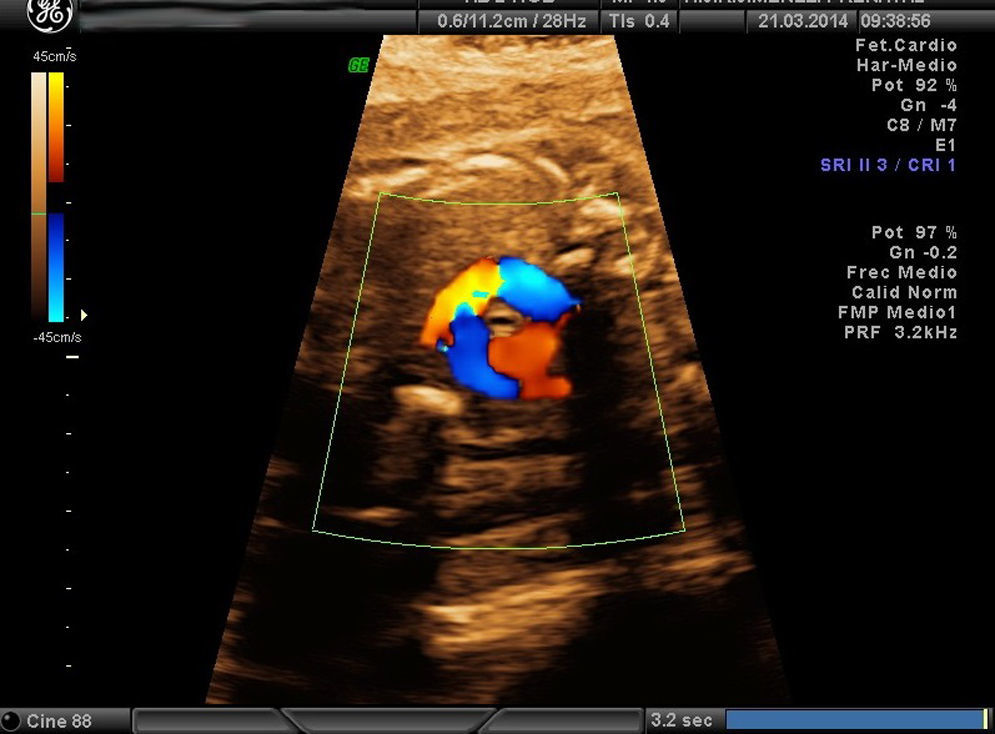
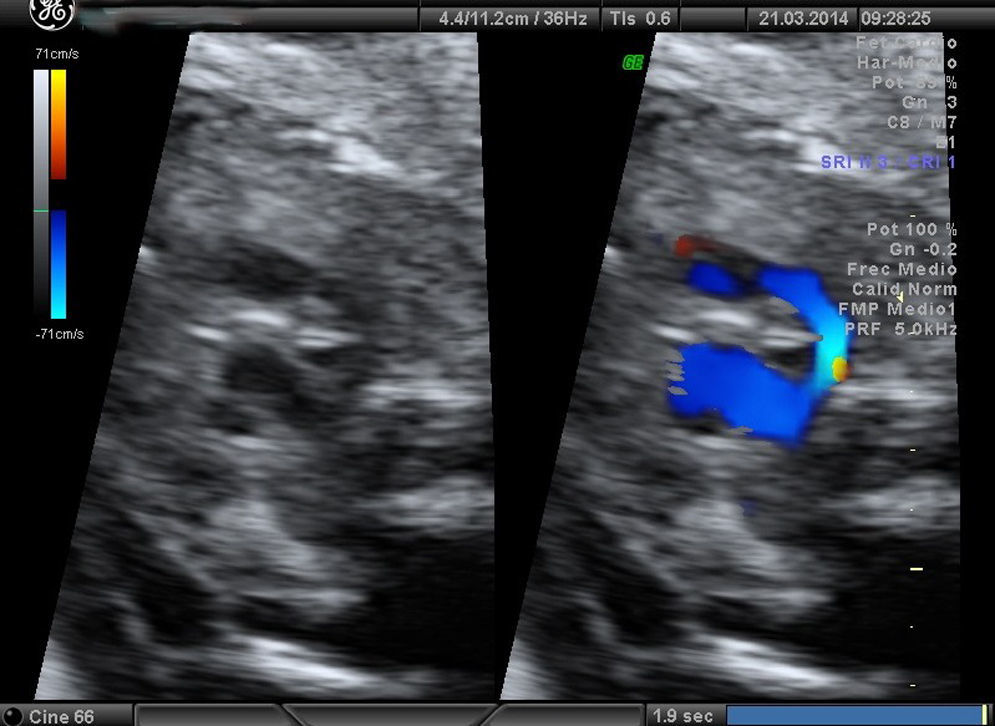
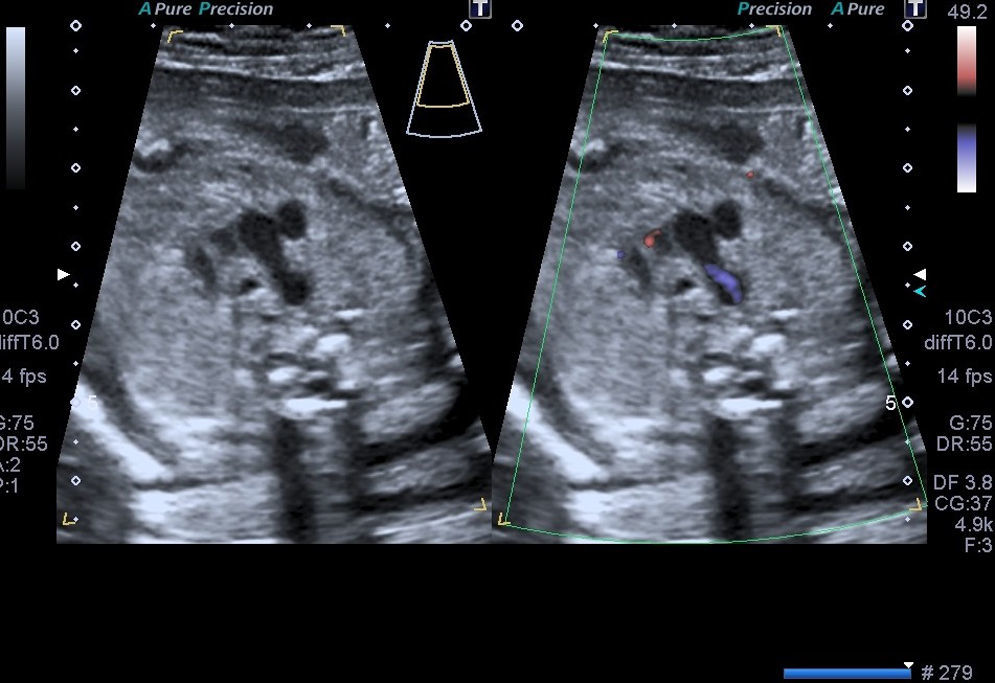
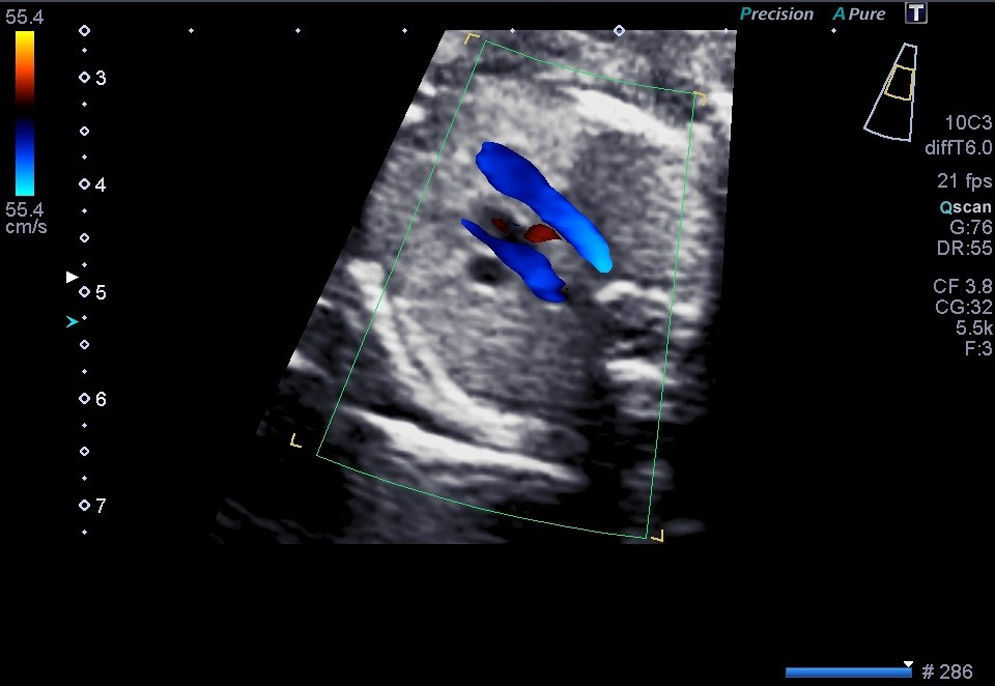
Casos clínicosCaso 1Paciente de 39 años, tercigesta y con antecedentes personales de talasemia minor y cirugía previa de mioma subseroso. Consulta por primera vez en nuestro servicio en la semana 12 del embarazo, con cribado combinado de cromosomopatías de bajo riesgo. Durante la práctica de la ecografía morfológica se detecta una sospecha de malformación cardíaca; el resto de las estructuras eran normales. Se visualiza un corte de 4 cámaras normal, con los tractos de salida pulmonar y aorta cruzados correctamente. En el corte «3VT» del mediastino superior descrito por Yagel se aprecia que la aorta pasa por el lado derecho de la tráquea y no por el izquierdo, como sería la norma. Se observa con claridad un anillo vascular alrededor de la tráquea que forma una imagen en «U» y no la característica «V» que dibujan normalmente la aorta y el ductus arterioso. Es esta imagen en «U» anómala que nos orienta hacia el diagnóstico del tipo de AAD, que correspondía probablemente a un AAD con arteria subclavia izquierda aberrante, en la que el anillo que rodea la tráquea está formado por la aorta ascendente, el AAD (a la derecha de la tráquea) (figs. 1-3) la arteria subclavia izquierda aberrante por detrás de la tráquea, y el ductus arterioso y el tronco de la pulmonar a la izquierda de esta. La aorta descendente se localiza a la izquierda de la columna vertebral y la cava inferior no presenta alteraciones. Se visualiza el timo dentro de parámetros normales. Ante la sospecha de AAD se aconseja la práctica de una amniocentesis y el estudio de la microdeleción del cromosoma 22, que no se realizaron en este caso. Se controla la gestación cada 2-3 semanas. En la semana 26 presenta alteración en el doppler de las arterias uterinas (>p95). Buen crecimiento fetal hasta la semana 32, que se correlaciona con una enfermedad hipertensiva del embarazo. La gestación finaliza a las 32+5 semanas, con maduración pulmonar completa, mediante una cesárea urgente por preeclampsia grave refractaria. Nace una mujer de 1.703g y Apgar 5-8, que precisa reanimación tipo ii. Se confirma una restricción del crecimiento intrauterino (p5) posnatalmente. La recién nacida ingresa desde quirófano por prematuridad y distrés respiratorio, confirmando mediante ecocardiografía fetal el diagnóstico prenatal completo. La madre precisó ingreso en la UCI, con una buena evolución posterior. Actualmente permanece sana a los 6 meses de vida.
Caso 2


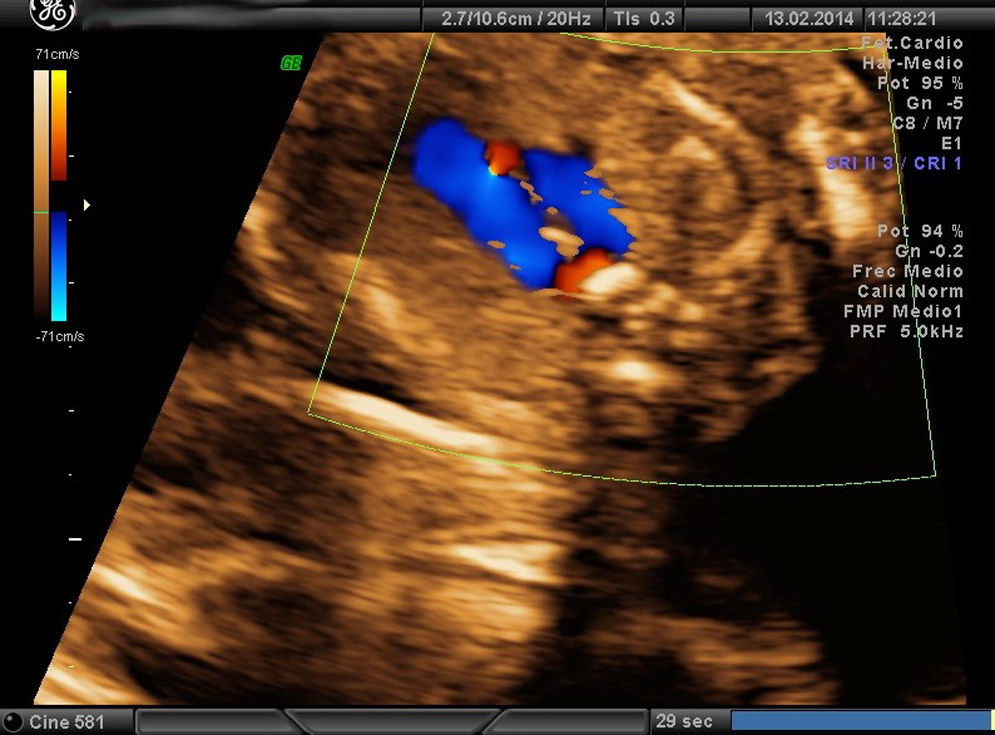
Paciente de 25 años, con el antecedente de interés de una tetralogía de Fallot intervenida a los 3 años. Secundípara. La primera visita de embarazo fue en la semana 12, con cribado combinado de bajo riesgo. En la ecografía morfológica se detecta la sospecha de una CC (corte de la «V» anómalo: tráquea a la izquierda de la aorta y ductus) (figs. 4 y 5) y el resto de las estructuras fetales eran normales. Se solicitó la valoración del CATCH-22 mediante amniocentesis, que resultó negativa. Técnica de diagnóstico genético. En esa misma semana se confirmó la sospecha diagnóstica de AAD. Corazón en situs solitus, eje de 45°, tamaño normal, corte de 4 cámaras normal, salida de grandes vasos cruzada. En el corte de mediastino alto se aprecia que la aorta ascendente pasa por la derecha de la tráquea, con visualización del ductus también a la derecha. Finaliza la gestación a las 39 semanas con un parto de inicio espontáneo y final eutócico, naciendo una mujer de 3.360g y Apgar 9/10. En el posparto se confirma el diagnóstico de AAD mediante ecocardiografía posnatal. Tras 5 meses de vida no presenta complicaciones clínicas.
Discusión

Las CC representan las malformaciones congénitas severas más frecuentes. Afectan aproximadamente a 8 por 1.000 recién nacidos y la mitad de ellos corresponde a malformaciones graves que, a pesar de los avances en cirugía cardíaca, siguen presentando una morbimortalidad no desdeñable. La mortalidad global alcanza el 25-30%, y un 50% llegará tan solo a la etapa de la adolescencia. Presentan una elevada relación con cromosomopatías, sobre todo si se asocian a otras malformaciones, y llegan al 15-25% si se presentan aisladas. Las anomalías del arco aórtico pueden pasar inadvertidas si se valora exclusivamente un corte de 4 cámaras (sensibilidad 40%) y las salidas de los grandes vasos, incluso en el corte de los 3 vasos (70%) descritos por Yoo et al.7. Sin embargo, si incluimos el estudio de los 5 planos de Yagel (90%)8, la probabilidad de apreciar estas anomalías es alta, sobre todo si se piensa en su posible existencia. La somnoluscencia de la tráquea durante el embarazo, ya que está repleta de líquido, permite su correcta visualización, lo que permite localizarla y detallar las relaciones que presenta con los grandes vasos en el mediastino superior. Precisamente esta es la base de la descripción que Yagel et al.8 realizan en el v plano. El autor empieza el estudio cardíaco con un corte de abdomen superior (i plano), y al trasladar el transductor del ecógrafo en sentido craneal, obtiene el corte de 4 cámaras (ii plano) y, posteriormente, la salida de la arteria pulmonar en el ventrículo derecho (iv plano), intercalando entre estos 2 cortes la visualización de la salida de la aorta (iii plano), que precisa de una pequeña angulación de la sonda hacia el hombro derecho del feto. El v plano se obtiene con la misma traslación paralela del i, ii y iv, y nos permite visualizar el llamado corte «3VT», donde podemos valorar la posición del arco aórtico. Es un método reproducible en casi todas las pacientes, en el que además de estudiar los cortes anteriormente descritos se valoran los arcos (aórtico y pulmonar) con un menor tiempo de exploración que cuando estos se evalúan mediante cortes paraesternales largos. La innovación de este método no solo radica en su reproducibilidad, sino en que por primera vez nos permite valorar la relación de los grandes vasos en el mediastino alto, de forma que se consiguen estudiar anomalías cardíacas, como las alteraciones del arco aórtico, que hasta ahora no podían apreciarse en el período prenatal. En la figura del corte de los «3V» normal (fig. 6) puede apreciarse cómo se relacionan entre sí los grandes vasos y los demás órganos en el mediastino superior. El arco del ductus (pulmonar) se sitúa a la izquierda del tórax; adyacente a él y pegado a su derecha se ve el arco aórtico (aorta ascendente y descendente), que confluye con el ductus en la parte posterior y cierra la imagen característica en «V». Más a la derecha y en posición anterior se aprecia un corte coronal de la vena cava superior, mientras que por detrás de ella y a la derecha de la aorta se sitúa la tráquea, lo que configura la descrita imagen «3VT». Así, los casos que se describen en el artículo presentan el signo patognomónico para el diagnóstico de AAD, es decir, encontrar distantes entre sí el arco del ductus y el de la aorta en el corte coronal del mediastino alto, pudiendo ver entre ellos la tráquea, con aspecto sonoluscente, que los separa.

A partir de aquí se podrá intentar precisar la variedad de AAD detectada. Fundamentalmente existen 2 variedades de AAD: con arteria subclavia izquierda aberrante (o innominada) o con las ramas en espejo. La primera se caracteriza porque la primera rama que surge de la aorta es la carótida izquierda, seguida de la carótida derecha, la subclavia derecha y finalmente la subclavia izquierda, que surge desde la aorta descendente pasando por detrás del esófago y de la tráquea. Entre un 10 y un 50% se asocian a CC. Ecográficamente, se traduce en:
- -
Un anillo vascular que rodea la tráquea y el esófago; la aorta ascendente, el AAD a la derecha de la tráquea, la arteria subclavia izquierda aberrante por detrás, el ductus (izquierdo), el tronco de la pulmonar por la izquierda y el corazón delante (fig. 2), y ofrece una imagen en «U» vascular en vez de la conocida forma en «V».
- -
En un corte coronal de la tráquea y los bronquios se aprecia la vena cava y el arco aórtico a la derecha, mientras la pulmonar aparece a la izquierda.
- -
Si se utiliza el doppler color se aprecia un flujo de sangre anterógrada en el ductus y un llenado retrógrado desde este hacia la aorta descendente. Esta zona corresponde a la conocida como divertículo de Kommerell, descrito por Burckhard F. Kommerell en 19369.
En el caso del AAD con ramas en espejo, como indica su nombre, el arco de la aorta pasa por la derecha de la tráquea y la disposición de los troncos supracervicales es especular a la norma. De esta forma nos encontramos con un primer tronco que corresponde a la arteria innominada izquierda, de la cual emergen la carótida y la subclavia izquierdas; seguidamente aparece la carótida derecha y, finalmente, la subclavia derecha. El ductus suele unirse a la innominada izquierda de manera que no se junta con la aorta. Por ello, no se visualiza un anillo vascular como sucede en la primera anomalía. Con frecuencia se asocia a CC (tetralogía de Fallot), asociación que se conoce como enfermedad de Corvisart. Ecográficamente se caracteriza por:
- -
No se visualiza un anillo vascular (por lo ya comentado anteriormente).
- -
No se aprecia la imagen característica en «V» entre aorta y pulmonar, así como tampoco la imagen en «U» del caso previo, ya que el ductus izquierdo se forma con la arteria innominada izquierda y la pulmonar.
- -
La aorta descendente se encuentra desplazada a la derecha de la línea media que delimita la columna vertebral.
La posibilidad de diagnóstico prenatal de estas anomalías tiene un valor trascendental en 2 aspectos: el primero consiste en que, como alteración conotruncal, ya se presente asociada o no a otras CC, puede darse en relación con cromosomopatías, de las que destaca la microdeleción del cromosoma 22. Así, en la bibliografía10 se encuentra como en el 46% de los niños con AAD presentaban una microdeleción del cromosoma 22. En segundo lugar merece destacarse la repercusión clínica. La disposición del arco aórtico y sus ramas puede favorecer la clínica respiratoria (disnea) o digestiva (disfagia). En principio, y una vez descartadas las cromosomopatías, se trata de una malformación que no suele presentar complicaciones prenatales, aunque se desconozca, hasta el nacimiento, si existirá repercusión sobre las vías respiratoria o digestiva. Así, en la serie más amplia de diagnóstico prenatal presentada, solo un feto entre 18 presentó clínica11. Sin embargo, estos pacientes no están exentos de complicaciones a lo largo de su vida. Hasta un 75% de los individuos con clínica la presentarán en edad infantil, aunque las complicaciones puedan aparecer incluso en adultos12. No es infrecuente diagnosticar la anomalía tras procesos disneicos refractarios a los tratamientos habituales13 o como complicaciones vasculares, habitualmente en pacientes que desarrollan un aneurisma en el divertículo de Kommerell en el caso de AAD con arteria subclavia aberrante12.
En conclusión, creemos que el diagnóstico de estas alteraciones es factible dada la colaboración inestimable de la visualización de la tráquea, que permite ver la relación con los grandes vasos. Ello orientará sobre la necesidad de realizar la valoración de la dotación cromosómica del feto y permitirá discriminar a los pacientes que deberán controlarse durante su vida ante la posibilidad de complicaciones severas, no exentas de una morbimortalidad elevada.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
