









El cáncer de mama es una de las enfermedades malignas más frecuentes en las mujeres. Su incidencia se ha ido incrementando cada año hasta considerarse un problema sanitario de características epidémicas. Alrededor de un 15% de las mujeres desarrolla cáncer de mama a lo largo de su vida. En los últimos años se ha realizado una gran cantidad de trabajos sobre los factores de riesgo y pronósticos de esta enfermedad. En esta revisión de la bibliografía científica, se presenta el estado actual de los factores pronósticos relacionados con la regulación del ciclo celular en el cáncer de mama.
Breast cancer is one of the most common malignancies among women. The incidence of this disease has been increasing yearly and it could now be considered an epidemic disease. Approximately 15% of women will develop this kind cancer during their lifetime. In the last few years, multiple studies have been carried out on the risk and prognostic factors of breast cancer. We provide a review of the scientific literature on the current situation of molecular prognostic factors for breast cancer related to cell cycle control.
El cáncer de mama es la enfermedad maligna más frecuente en las mujeres de todo el mundo1,2. Su incidencia se ha ido incrementando en todos los países occidentales3,4. En la actualidad puede considerarse un problema sanitario de características epidémicas, ya que se estima que de 1 a 2 mujeres de cada 10 desarrollará cáncer de mama a lo largo de su vida5–9. Además, es la primera causa de muerte entre las mujeres a escala mundial10. En España, la incidencia asciende a 16.000 nuevos casos anuales2–4,11. Esto supone casi un 10% del total de diagnósticos de cáncer en este país.
En los últimos años, el aumento del volumen de estudios, de ensayos clínicos y de publicaciones relacionados con la etiología, la biología molecular, el diagnóstico y el tratamiento del cáncer de mama es evidente. Esto implica un aumento en las expectativas de alcanzar una mayor comprensión de la historia natural y del comportamiento clínico de la enfermedad y, por tanto, lograr una disminución de las tasas de mortalidad por esta enfermedad.
El estudio de los diferentes factores moleculares implicados en el ciclo celular ha supuesto en los últimos años un avance en el conocimiento del comportamiento intrínseco de las células cancerígenas mamarias. Esto posibilita orientar un pronóstico distinto para cada tumor, lo que influye en su tratamiento y en la actitud diagnóstico-terapéutica posterior. En la actualidad, se tiende a agrupar los factores pronósticos moleculares y a analizar una gran cantidad de éstos simultáneamente a fin de dar un perfil molecular de buen o mal pronóstico, hecho que parece ser más efectivo y fiable que las clasificaciones pronósticas clásicas de St. Gallen, Nottingham y las de la NIH12–15.
Factores pronósticos molecularesEntre los distintos pasos que realiza una célula normal para convertirse en una célula tumoral, el primero es proliferar excesivamente y evitar los mecanismos de control del ciclo celular que actúan fisiológicamente para impedir esto.
Para esto, deben activarse los oncogenes (a partir de genes normales implicados en la proliferación celular denominados protooncogenes) y deben perder su actividad los denominados genes oncosupresores, que ejercen una acción fisiológica de freno sobre la proliferación. La alteración de esta situación fisiológica de equilibrio entre protooncogenes y genes oncosupresores es tan importante, que muchas de las proteínas implicadas en esta interrelación son algunos de los más valiosos marcadores pronósticos moleculares del cáncer de mama.
A continuación se revisarán los factores pronósticos moleculares más relevantes relacionados con el ciclo celular.
Ploidía y actividad proliferativaLas células normales presentan un contenido de ácido desoxirribonucleico (ADN) y una organización cromosómica estable, mientras que las células cancerosas tienen la propiedad de la variedad genética, tanto molecular como estructuralmente. El estudio del ciclo celular mediante citometría de flujo ha permitido caracterizar alguna de estas diferencias16. A causa de lo anterior es bien sabido que la ploidía, entendida como el contenido de ADN en el núcleo celular y la cantidad de células en fase de síntesis de ADN (fase de síntesis [S]) del tumor, se correlaciona significativamente con el grado histológico y el contenido de receptores hormonales del tumor17.
Además, la aneuploidía (fig. 1) se asocia a ganglios axilares invadidos, menor intervalo libre de enfermedad y menor supervivencia global; mientras que los diploides se asocian a bajos valores de la fase S y presentan un buen pronóstico18,19. Se ha descrito una relación inversa entre la aneuploidía y las concentraciones de estrógenos, de modo que los tumores aneuploides frecuentemente no expresan los receptores estrogénicos (RE), al igual que ocurre con los receptores de la progesterona20.
, con un solo pico en G0 y G1 y aneuploide (derecha) con 2 picos que representan 2 poblaciones celulares.")
Con respecto a la fase S, cuando ésta es elevada, los tumores suelen ser escasamente diferenciados, negativos para la expresión de receptores hormonales y tienen un menor intervalo libre de enfermedad y menor supervivencia global, como ocurre con la aneuploidía; asimismo, muchos autores lo consideran un factor pronóstico independiente21. La fase S del ciclo celular y la expresión del antígeno Ki-67 tienen una misma significación clínica en los tumores de mama: la proliferación. Sin embargo, los resultados no son completamente superponibles16.
Gen c-mycEl c-myc es un factor de trascripción implicado en la regulación de todas las actividades celulares, incluidas la proliferación, la diferenciación y la apoptosis22. El gen c-myc, localizado en el cromosoma 8q24, se amplifica aproximadamente en un 20 a un 30% de los cánceres de mama y se correlaciona poco con la expresión del c-myc, tanto en el ácido ribonucleico mensajero como en las proteínas; la amplificación del c-myc suele ocurrir más en el carcinoma ductal infiltrante que en el carcinoma in situ23.
Es interesante resaltar que, mientras las alteraciones del c-myc están presentes en determinados cánceres mamarios primarios, éstas no siempre se detectan en las metástasis ganglionares linfáticas correspondientes (aunque haya muchos autores que defienden su implicación principal en la tumorgénesis23), lo que indica su papel potencial en la iniciación y promoción anteriores a la invasión24.
La amplificación del c-myc también se asocia a tumores más agresivos y es un factor de mal pronóstico independiente en cáncer de mama, ya que se relaciona con un intervalo libre de enfermedad y una supervivencia global disminuida, cuyo efecto es aditivo al de otros factores de mal pronóstico25,26.
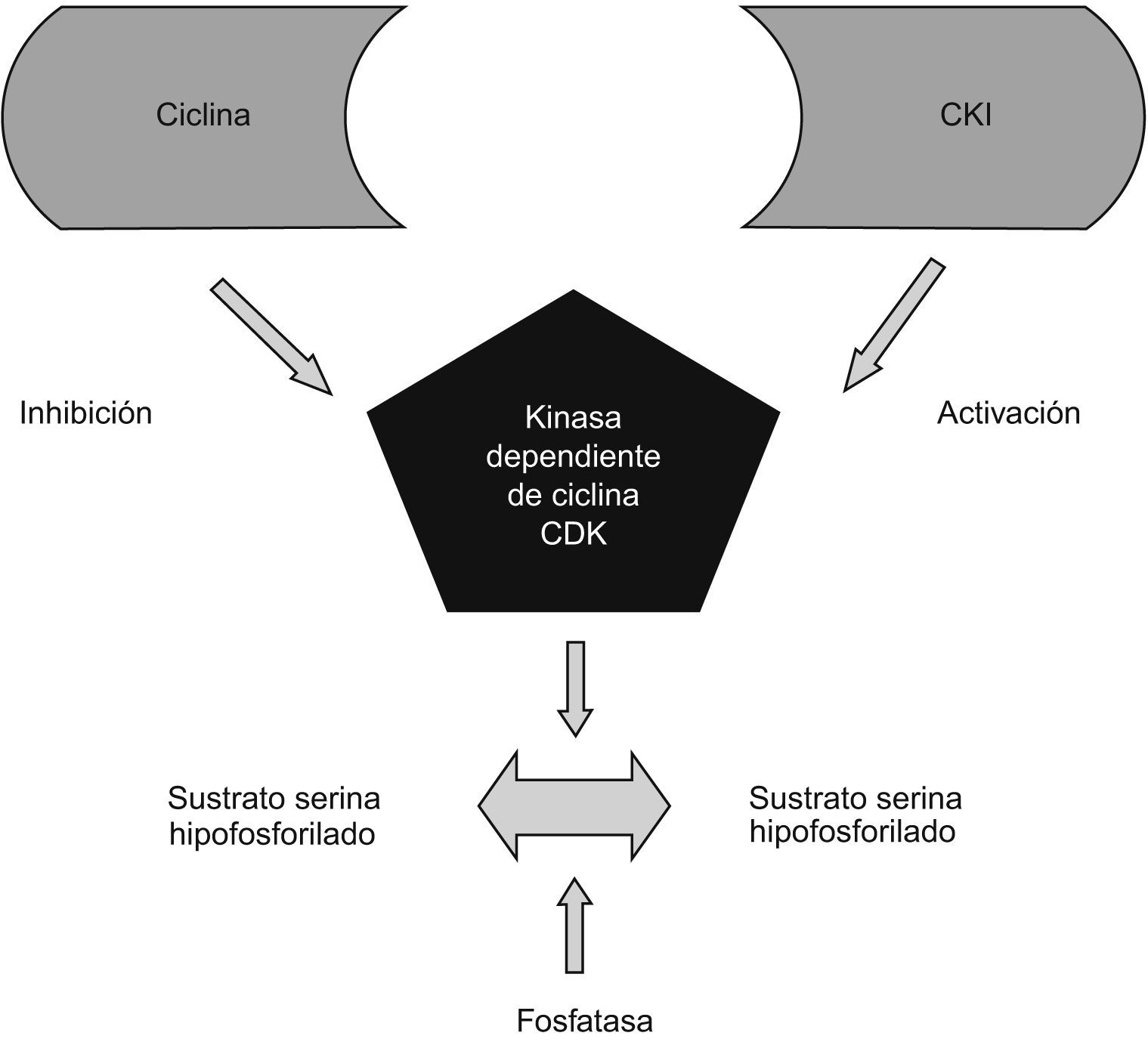
Sistema de cinasa dependiente de ciclinaLos mecanismos moleculares básicos que controlan el paso de la célula a través de las distintas etapas del ciclo celular dependen de una serie de holoenzimas, constituidas por una subunidad reguladora llamada ciclina y por una subunidad catalítica llamada CDK (cyclin dependent kinase ‘cinasa dependiente de ciclina’). La función principal de esta enzima es inactivar los mecanismos que impiden la progresión de la célula hacia la división en sus distintas fases. La distintas ciclinas se involucran de modo diferente en cada parte del ciclo celular (fig. 2).

Las alteraciones del punto de control del ciclo celular de la fase G1 (gap 1 ‘intervalo 1’) a la fase S parecen estar implicadas en el cáncer de mama humano. En su estado natural, el complejo de CDK, con varias ciclinas activadas de forma secuencial, inicia la progresión hacia la fase S a través de la fosforilación de la proteína pRB y la liberación del factor de trascripción E2F que, a su vez, activa los genes responsables de la replicación del ADN. Además, hay una variedad de CKI (cyclin dependent kinase inhibitors ‘inhibidores de las CDK’) que compiten con las ciclinas en la unión a las CDK para impedir la progresión a través de las fases G1 a S. De esta forma, algunas CDK, unidas a sus inhibidores, podrían tener alguna función como oncosupresores27,28 (fig. 3).

De entre las CDK, las más importantes son la CDK2, CDK4 y CDK6, que son las responsables de unirse a las ciclinas en las distintas fases del ciclo celular.
También cabe destacar, de toda la familia de la ciclinas, a la ciclina D1 y a la ciclina E. La proteína ciclina D1 se encuentra sobreexpresada en el 35 al 50% de cánceres de mama y controla la progresión del ciclo celular29. Algunos trabajos demuestran su asociación a positividad para los RE, lo que ciertos autores, a diferencia de otros, asocian a mal pronóstico. Lo que parece claro es que está implicada en varias etapas de la carcinogénesis y la progresión tumoral30. Con respecto a la ciclina E, se postula que es la causante de que la célula entre en fase S, su sobreexpresión (en el 25% de los cánceres de mama) no se ha asociado al riesgo de recidivas locorregionales ni metástasis31, pero sí se relaciona con los tumores mamarios más indiferenciados y podría aumentar la sensibilidad a determinados tratamientos quimioterapéuticos32.
Con respecto a las CKI, una de las familias más importantes incluye a las proteínas Ink4 (p15, p16, p18 y p19) que forman complejos con CDK4 y CDK6, así como con las ciclinas D, para detener la progresión33. Otro grupo está compuesto por las proteínas Cip o Kip que incluyen a p21, p27 y p57, y tienen un amplio margen de especificidad y un gran espectro de actividades inhibidoras. Algunas de estas CKI han estado implicadas en cáncer de mama; una de las proteínas Ink4 (p16) (fig. 4) y 2 de las proteínas Cip o Kip (p21 y p27).

Los estudios realizados hasta el momento sobre estos inhibidores revelan una alteración de estas proteínas en las células tumorales malignas, no homogénea en los numerosos estudios, que les hace perder su capacidad inhibitoria, proapoptótica y oncosupresora y que la caracteriza como un factor de mal pronóstico en cáncer de mama34–36.
Protooncogen C-erb-B2El c-erb-B2 (HER2/neu) es un protooncogen localizado en el cromosoma 17q21 que codifica un receptor de la tirosina cinasa transmembrana (ERB2 o p185), éste comparte una extensa similitud con el receptor del factor de crecimiento epidérmico. Su amplificación se detectó en un 20 a un 30% de los tumores invasivos de mama27. Es interesante resaltar que las alteraciones del c-erb-B2 se registraron en un 40 a un 60% de los casos de carcinoma ductal in situ, y están más extendidas en los subtipos comedo que en los cribiformes, lo que indica que juegan un papel en la progresión de las lesiones premalignas23.
Se ha relacionado la presencia de ERB2 en las protuberancias de la membrana plasmática necesarias para la motilidad celular, lo que juega un papel esencial en la invasión tumoral y metástasis. La sobreexpresión de c-erb-B2 se relaciona con peor pronóstico tumoral, menor intervalo libre de enfermedad y menor supervivencia global, presencia de metástasis en ganglios axilares, independencia de las hormonas, mayor capacidad proliferativa y resistencia al tamoxifeno. No se relaciona con la edad, grado histológico ni con el tamaño tumoral37–41. Cabe mencionar que la sobreexpresión del c-erb-B2 es variable en los distintos tipos tumorales, incluso su expresión es heterogénea dentro de un mismo tumor, hecho que suele estar subestimado y que puede afectar a su sensibilidad ante los distintos tratamientos disponibles, como el trastuzumab42.
RetinoblastomaEl gen RB, localizado en el cromosoma 13q14, codifica una proteína nuclear (pRB) que cuando se fosforila mediante complejos de CDK, libera E2F y de esta forma permite la progresión del ciclo celular hacia la fase S23. Se han encontrado alteraciones de esta región en el 30% de los cánceres de mama.
En diversos estudios parece ya demostrado que las concentraciones de pRB se relacionan con la actividad proliferativa tumoral, el tamaño, el grado histológico, el grado nuclear y los valores la expresión de receptores hormonales; sin embargo, no se relaciona con los valores de p5343–45.
Gen p53El gen p53 está localizado en el cromosoma humano 17p13 y codifica una fosfoproteína nuclear de 393 aminoácidos que funciona en el control del ciclo celular como mediadora de la diferenciación, de la reparación del ADN y de la apoptosis. Este gen se activa cuando la célula sufre daños en su ADN o cuando recibe estrés celular; bajo estas condiciones, el gen p53 sufre un proceso de activación postraduccional y muchos de sus residuos se modifican por fosforilación, acetilación o sumolización.
En situaciones normales, el gen p53 está presente con valores bajos en todas las células. Cuando agentes externos agreden a la célula, el gen p53 activa sus funciones que conllevan la parada del ciclo celular, la reparación del ADN por daño o la entrada en apoptosis. Estos procesos son en gran medida dependientes de la capacidad del gen p53 para activar la trascripción de determinados genes. Como tal, el gen p53 es un oncosupresor, es el gen somáticamente mutado más común en los cánceres esporádicos y, cuando es mutante en la línea germinal, hace que el locus de susceptibilidad del cáncer hereditario sea responsable del Síndrome de Li-Fraumeni22,46–48.
La proteína p53, denominada así por su peso molecular (53 KD), juega un papel crucial en la regulación de la proliferación celular y en la respuesta de la célula a estímulos de estrés. La proteína p53 induce tanto la parada del ciclo celular que previene la replicación del ADN dañado, como la activación de la apoptosis para eliminar células «defectuosas»49–52.
En el cáncer de mama humano, se han detectado alteraciones de p53 en las proteínas o en el ADN en un 25 a un 50% o en un 15 a un 35% de los casos, respectivamente. El estudio del espectro de mutaciones de p53 indica una fuerte presión selectiva para mutaciones localizadas predominantemente en el dominio de unión al ADN de la proteína51. Casi el 90% de las mutaciones presentes en tumores humanos se localizan entre los exones 4 a 9 correspondientes a la región de unión al ADN donde hay “puntos calientes” de mutación, que pueden causar en p53 tanto pérdida de la función supresora de tumores como ganancia de la función oncogénica, y que modifican el repertorio de genes que controla p53 como factor de trascripción53, lo que interrumpe el papel de p53 como regulador natural del crecimiento y contribuye así a la tumorogénesis.
Los genes que regula p53 trascripcionalmente (tabla 1) pueden agruparse de acuerdo con las funciones que realizan. Se ha encontrado que p53 regula genes implicados en la inhibición del ciclo celular, en la reparación del daño en el ADN, en la inhibición de la angiogénesis (formación de novo de vasos) y en la inducción de la apoptosis54. Además, p53 regula la trascripción de la proteína MDM2, que a su vez regula los valores de p53, lo que genera un sistema autoregulable. Por esto, la inusual función de p53 puede conducir a trastornos de la diferenciación y del control del ciclo celular mediante una interrupción del punto de control de la fase G1 a la fase S, que puede resultar en una pérdida de la respuesta apoptótica al daño del ADN y en la consiguiente replicación del ADN de un genoma sin reparar; es decir, que puede incrementar la inestabilidad genómica que a su vez facilita la aparición de mutaciones de otros genes22,46,48. Además, la alteración de p53 hará que se pierda el control sobre la angiogénesis, lo que favorecerá la progresión tumoral. Normalmente, la proteína p53 tiene una vida media muy corta y se halla en pequeñas cantidades en las células normales, pero al mutar, se alarga su vida media y se encuentra en las células tumorales en altas concentraciones.
Hay una serie de genes a los que p53 regula directamente y cuya función está directamente implicada en la apoptosis (tabla 1).
El más conocido es Bax, un miembro de la familia Bcl-2, que promueve la apoptosis celular y del que se hablará más adelante. De momento, cabe comentar que Bax está inducido por p53 aunque no en todos los tipos celulares. Se ha visto que las proteínas miembros de la familia de Bcl-2 funcionan, al menos en parte, regulando la liberación de citocromo C de la mitocondria, un suceso observable en la apoptosis dependiente e independiente de p53. El gen Bax tiene sitios de unión a p53 en su promotor y se regula positivamente en respuesta al daño en el ADN y en p53 en varios sistemas (fig. 5). La introducción de Bax en células resulta en una muerte celular rápida que se puede inhibir por coexpresión de Bcl-2 y Bcl-X. De manera similar, la muerte mediada por p53 puede ser inhibida por Bcl-2 y Bcl-X, lo que es consistente con un papel para Bax en la vía de muerte de p53. Sin embargo, Bax no está implicada siempre en la apoptosis que media p53 y no representa el único mecanismo de muerte celular dependiente de p5355–57.

Con respecto a la relación entre la sobreexpresión de p53 y otros factores clínicos y pronósticos, hay una gran variabilidad entre los distintos estudios, aunque parece haber acuerdo en que p53 es un factor pronóstico independiente que se relaciona con la intensidad tumoral y que se asocia a positividad para c-erb-B2, Ki-67 y RE. Además se ha observado que tiene una asociación significativa con el período libre de enfermedad y predice la respuesta tumoral al tamoxifeno58–61.
