




INTRODUCCIÓN
Los tumores desmoides (fibromatosis) son tumores benignos de origen mesenquimatoso. En la zona mamaria, tanto clínica como radiológicamente, pueden parecer un carcinoma; su diagnóstico definitivo es el anatomopatológico. Se caracterizan por presentar un comportamiento agresivo con alta tendencia a la recidiva local, pero sin tendencia a metastatizar a distancia. Presentamos un caso de fibromatosis mamaria en una paciente de 23 años con antecedente de tumorectomía mamaria ipsolateral, que tanto clínica como radiológicamente simulaba un carcinoma mamario.
CASO CLÍNICO
Paciente de 23 años, nuligesta, remitida a nuestra unidad de patología mamaria en enero de 2005, por tumoración mamaria izquierda.
No presenta historia familiar de carcinoma mamario ni antecedentes médicos de interés. Hacía 2 años había sido sometida a tumorectomía en cola de la mama izquierda por un nódulo mamario (el análisis anatomopatológico lo informó como fibroadenoma).
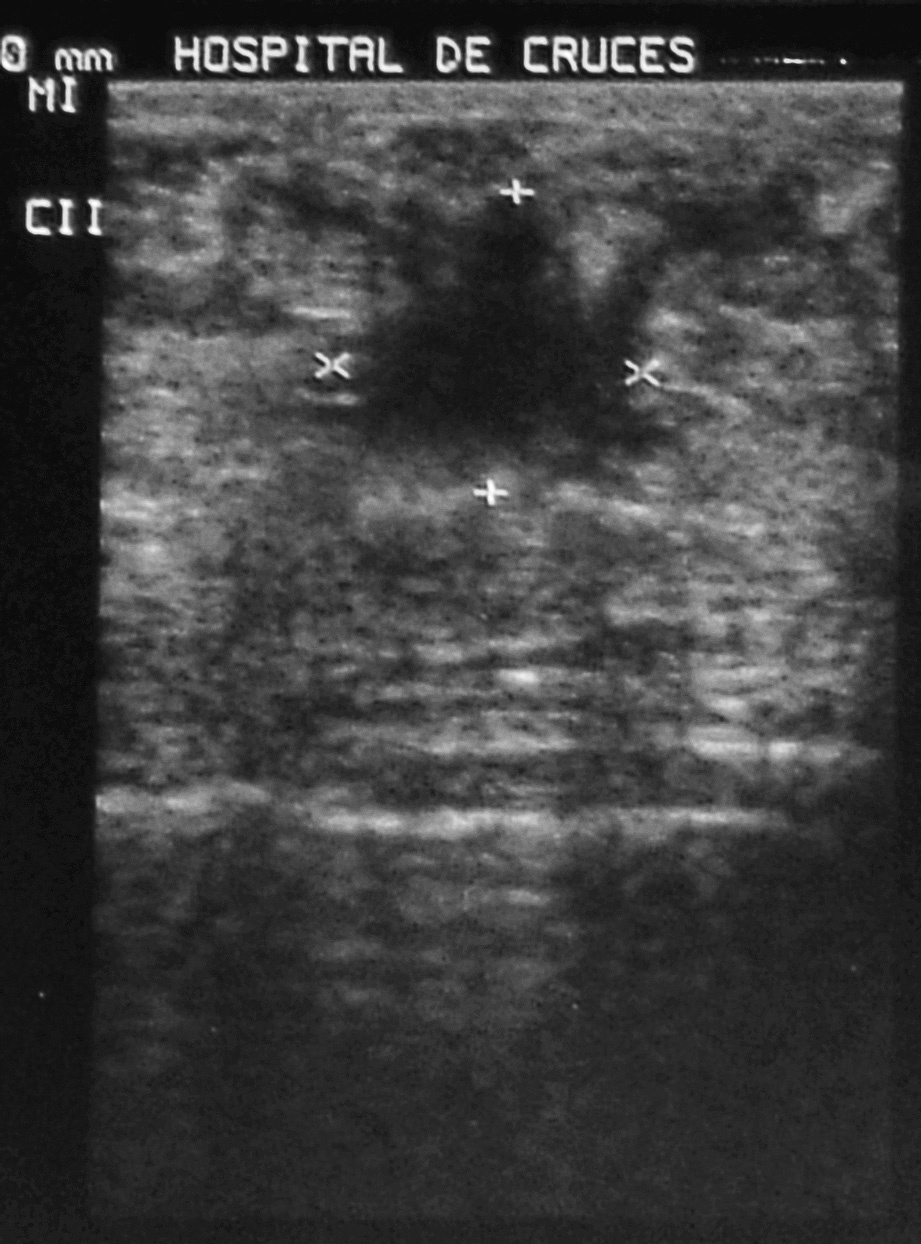
A la exploración mamaria se puso de manifiesto un aumento de densidad irregular en la unión de cuadrantes superiores (UCS) de la mama izquierda, indolora y sin retracción cutánea asociada. No se palpan adenopatías axilares ni supraclaviculares, ni se objetiva telorrea. En la ecografía mamaria se describen varios nódulos en ambas mamas, con características ecográficas de fibroadenomas, salvo un nódulo sólido en la región periareolar (UCS) de la mama izquierda, de bordes irregulares y ecogenicidad heterogénea, con zonas de atenuación posterior de 15 ƒ 14 mm, en relación con un posible carcinoma mamario (fig. 1).
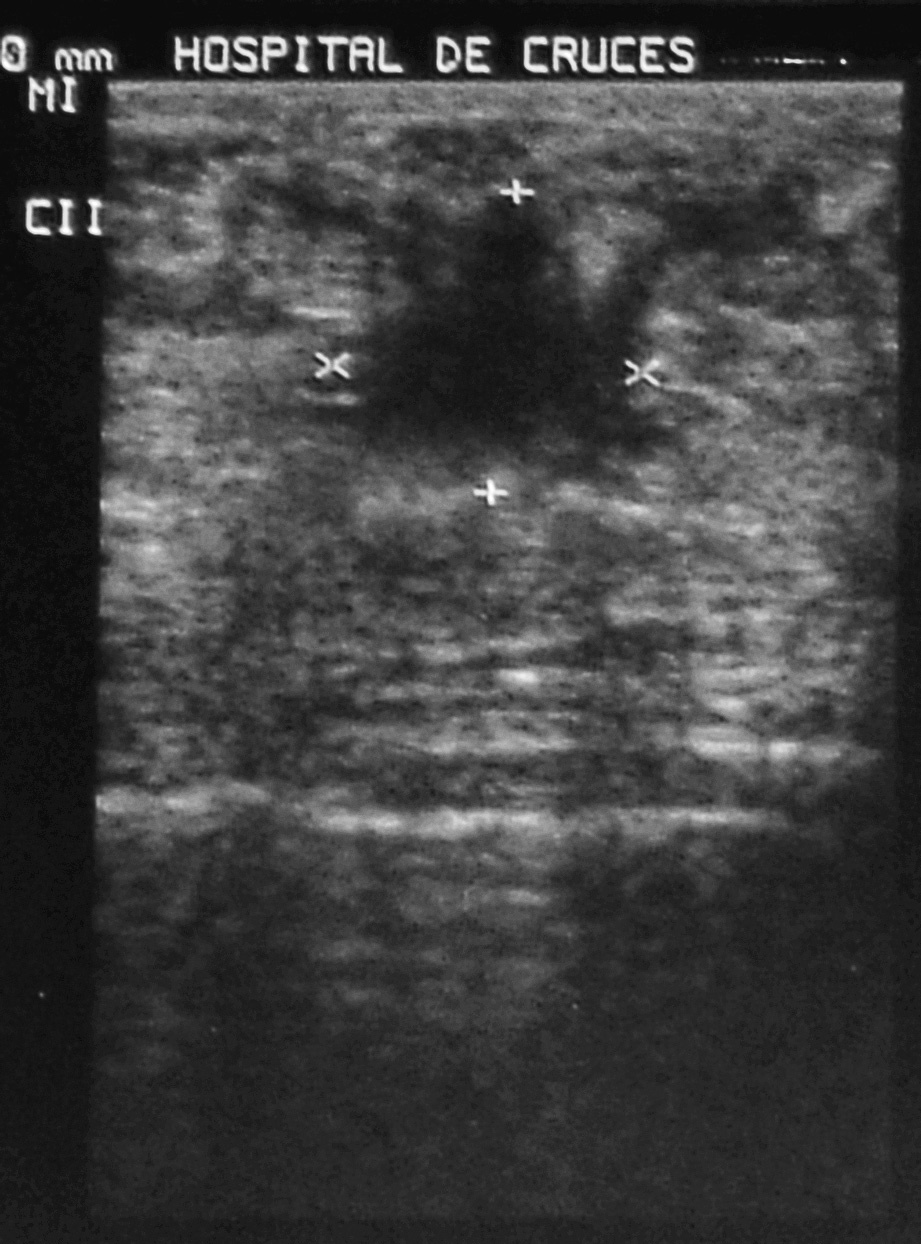
Fig. 1. Ecografía mamaria: nódulo de bordes irregulares y ecogenicidad heterogénea, con zonas de atenuación posterior.
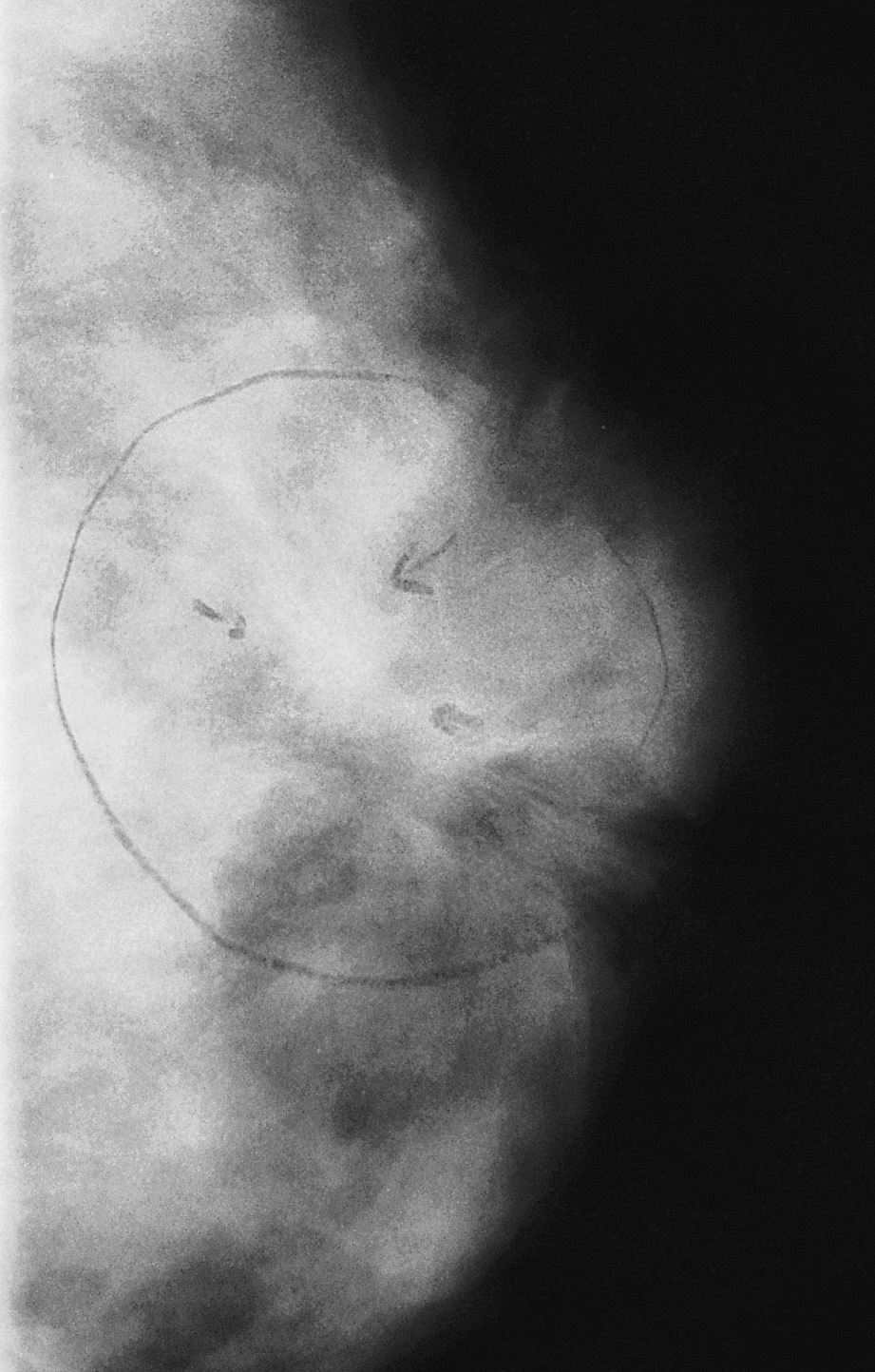
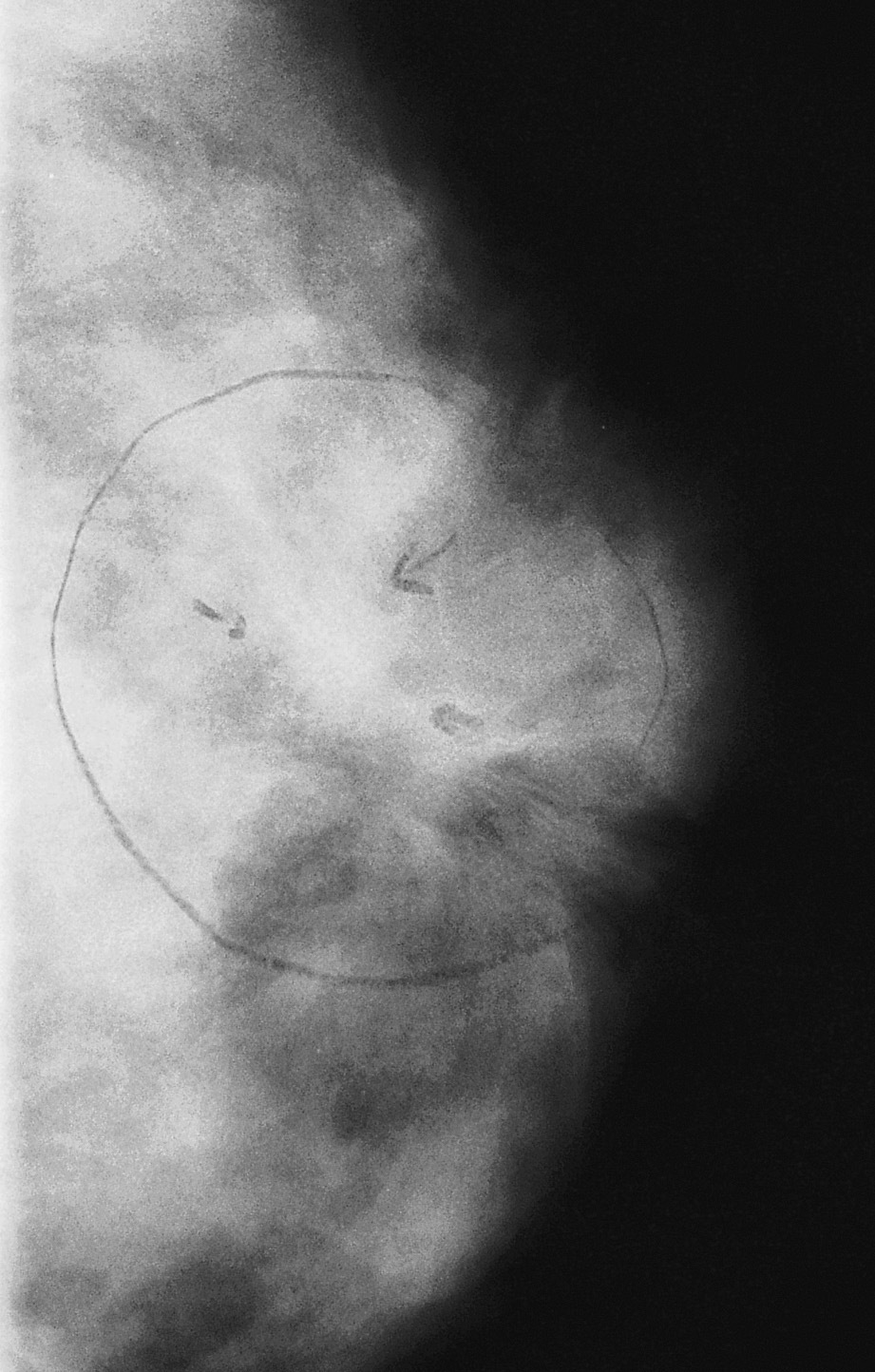
Fig. 2. Mamografía: nódulo estrellado, de bordes irregulares de 13ƒ10 mm de diámetro, compatible con carcinoma mamario.
La mamografía informó de imagen nodular en la región retroareolar de mama izquierda, estrellada, de bordes irregulares y espiculados, que retrae la piel supraadyacente, de 13 ƒ 10 mm, compatible con carcinoma mamario (fig. 2).
En la citología mediante punción-aspiración con aguja fina (PAAF) se observaron numerosos fibroblastos (en número superior a una reacción regenerativa) disociados o formando pequeños grupos, con núcleos ovalados, grandes, centrales, de cromatina blanda y pequeños nucléolos. El informe definitivo es de posible tumor de estroma, con sospecha de malignidad.
Se programó a la paciente para tumorectomía con estudio anatomopatológico intraoperatorio. Se realizó la resección del nódulo con márgenes de seguridad, alcanzando la aponeurosis del músculo pectoral. El estudio intraoperatorio fue informado de fibromatosis mamaria con bordes libres.
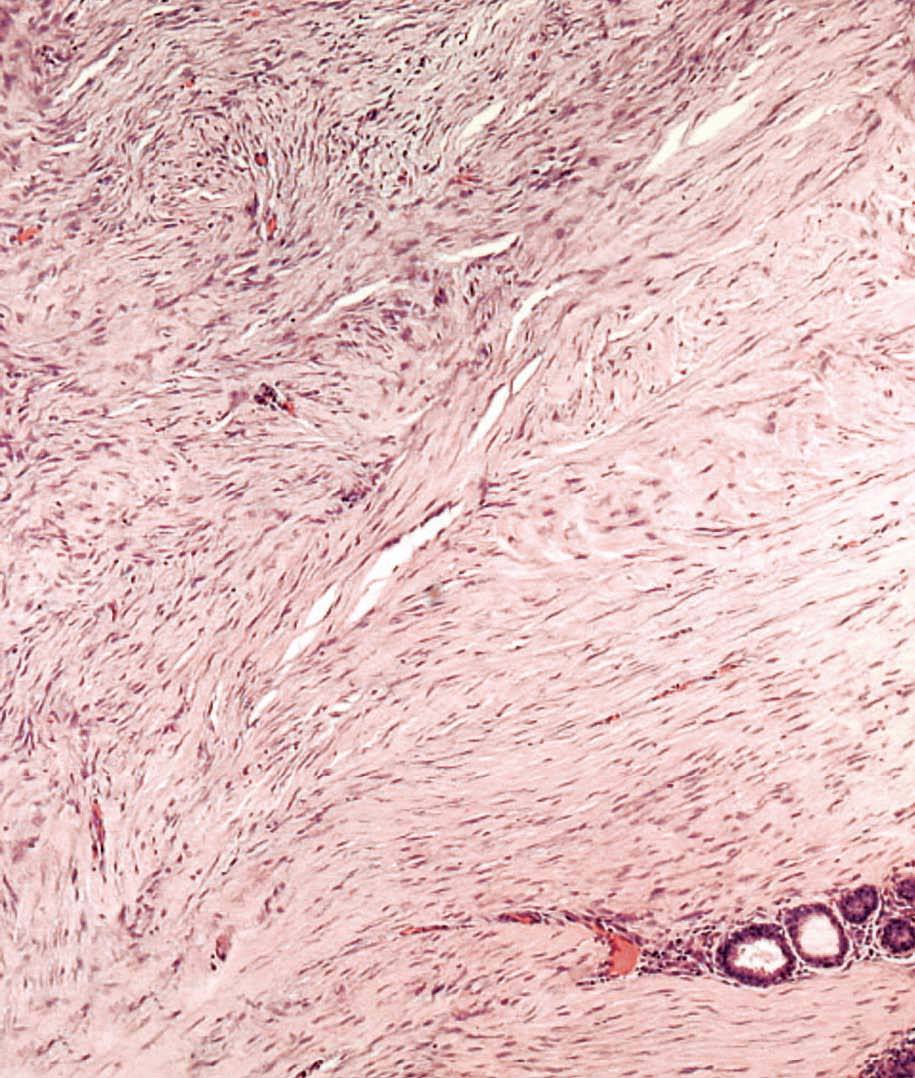
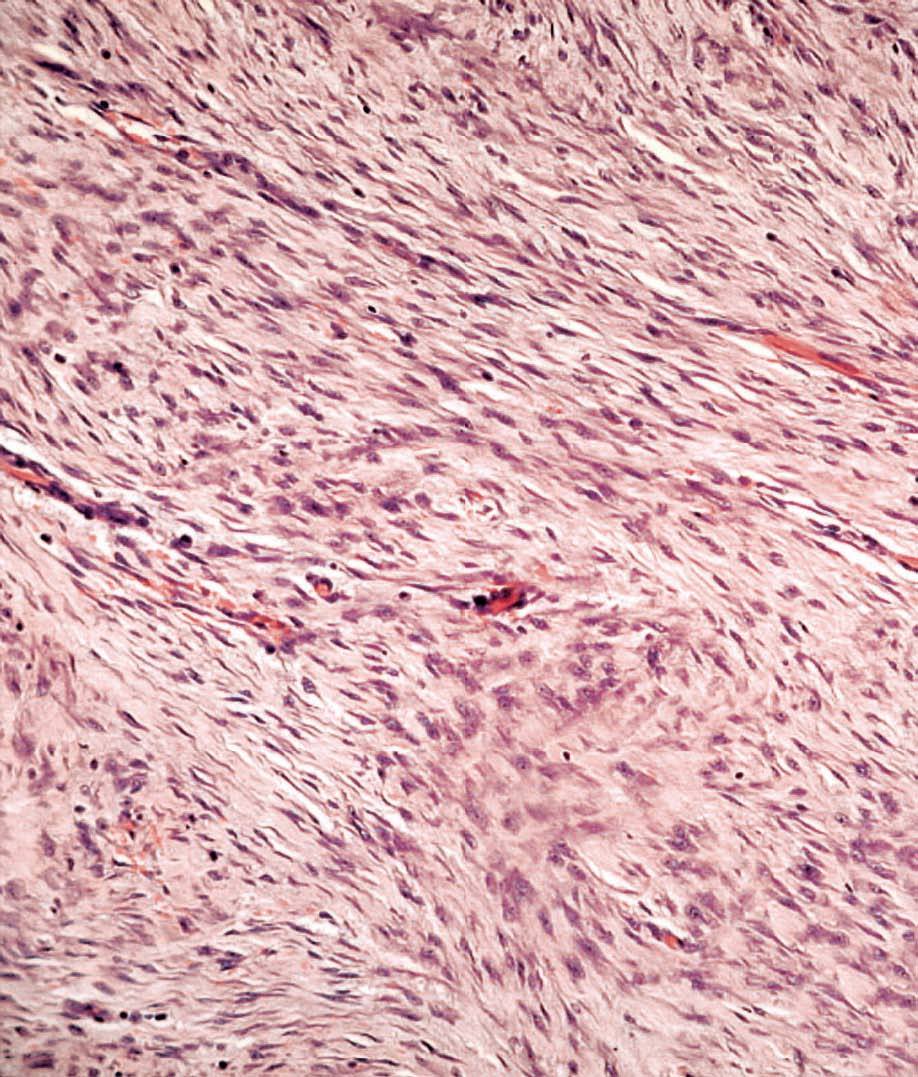
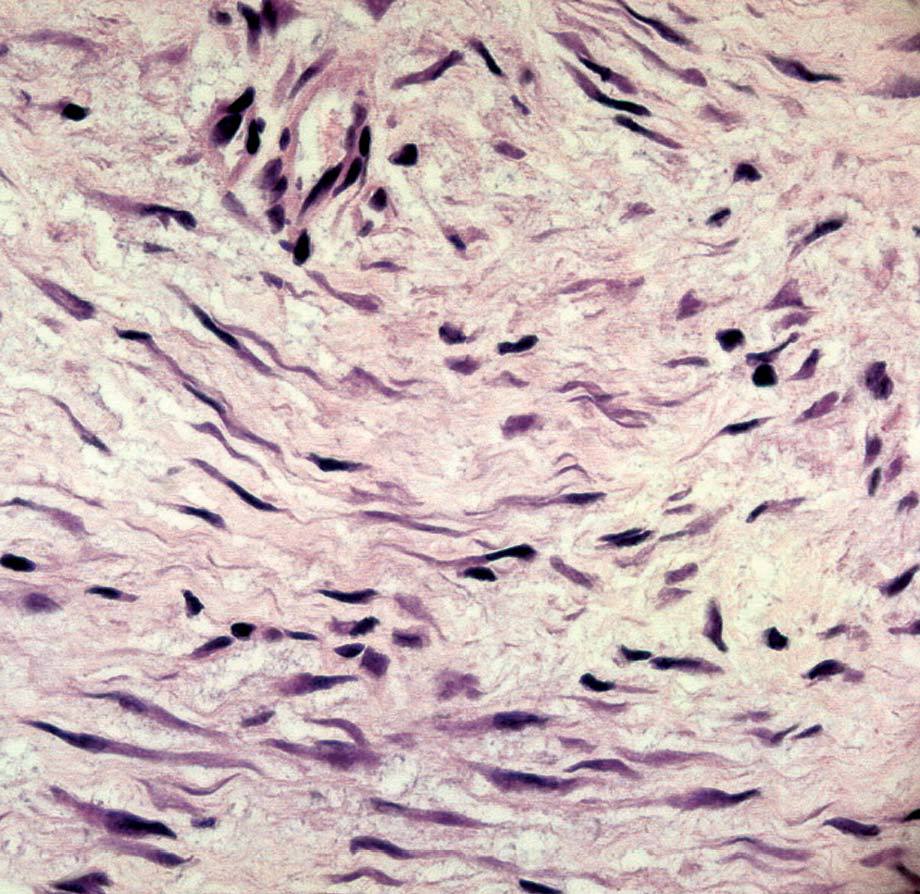
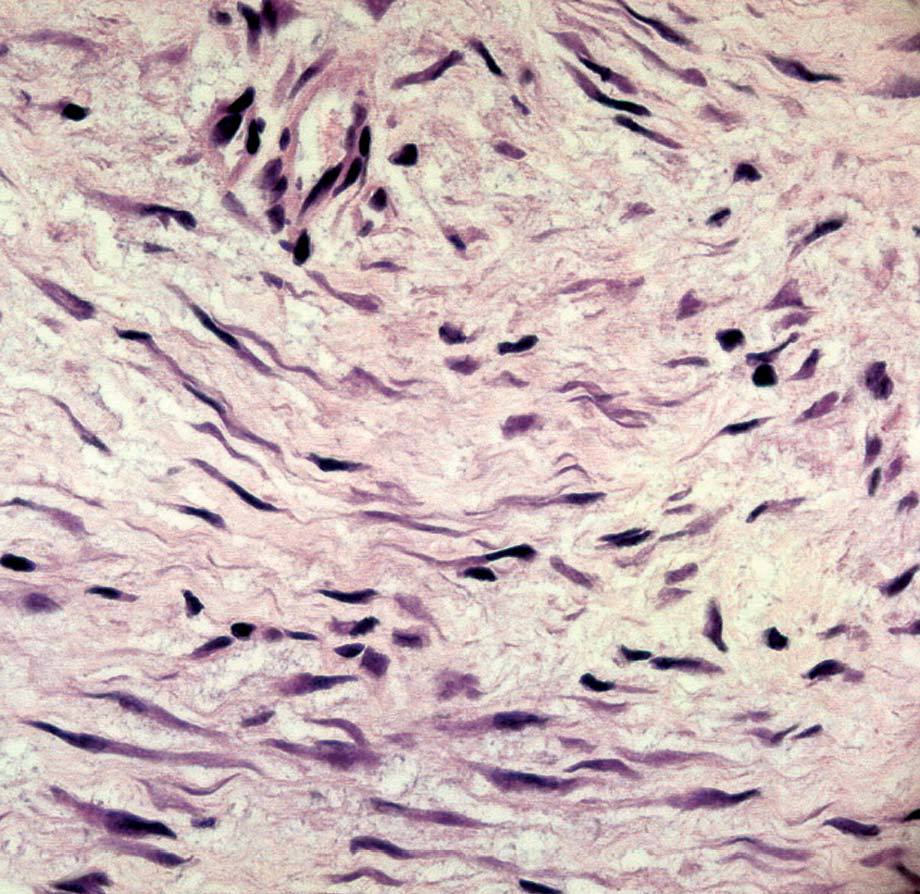
La anatomía patológica macroscópica informó de pieza de 6 ƒ 5 cm, en cuya sección se identifica una zona mal definida, de consistencia muy firme. El estudio microscópico muestra una lesión infiltrativa, de límites imprecisos, de estructura nodular y fasciculada, formada por células fusiformes de núcleos alargados sin signos de atipia, inmersas en una matriz colagénica densa (fig. 3). Aunque el índice proliferativo del tejido tumoral es bajo, el tumor engloba y comprime el parénquima mamario adyacente. Las células tumorales presentan una fuerte reactividad para la vimentina (fig. 4).
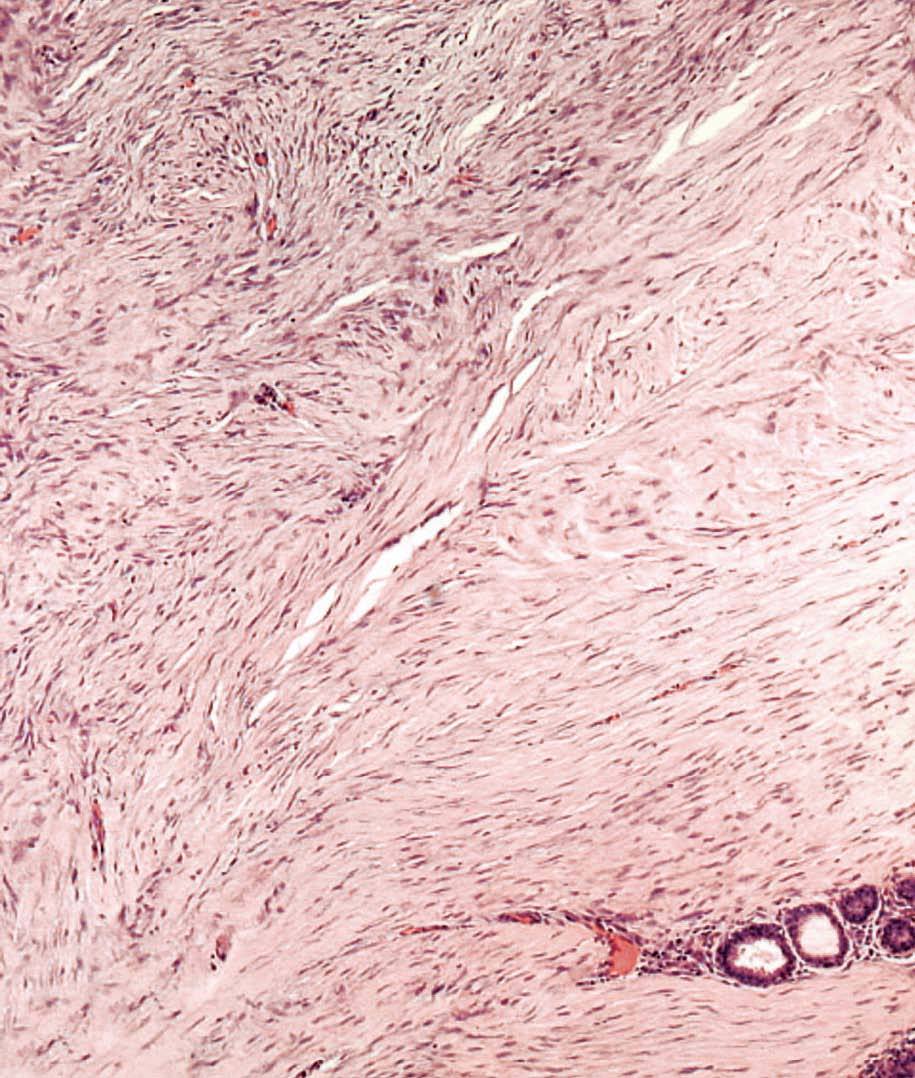
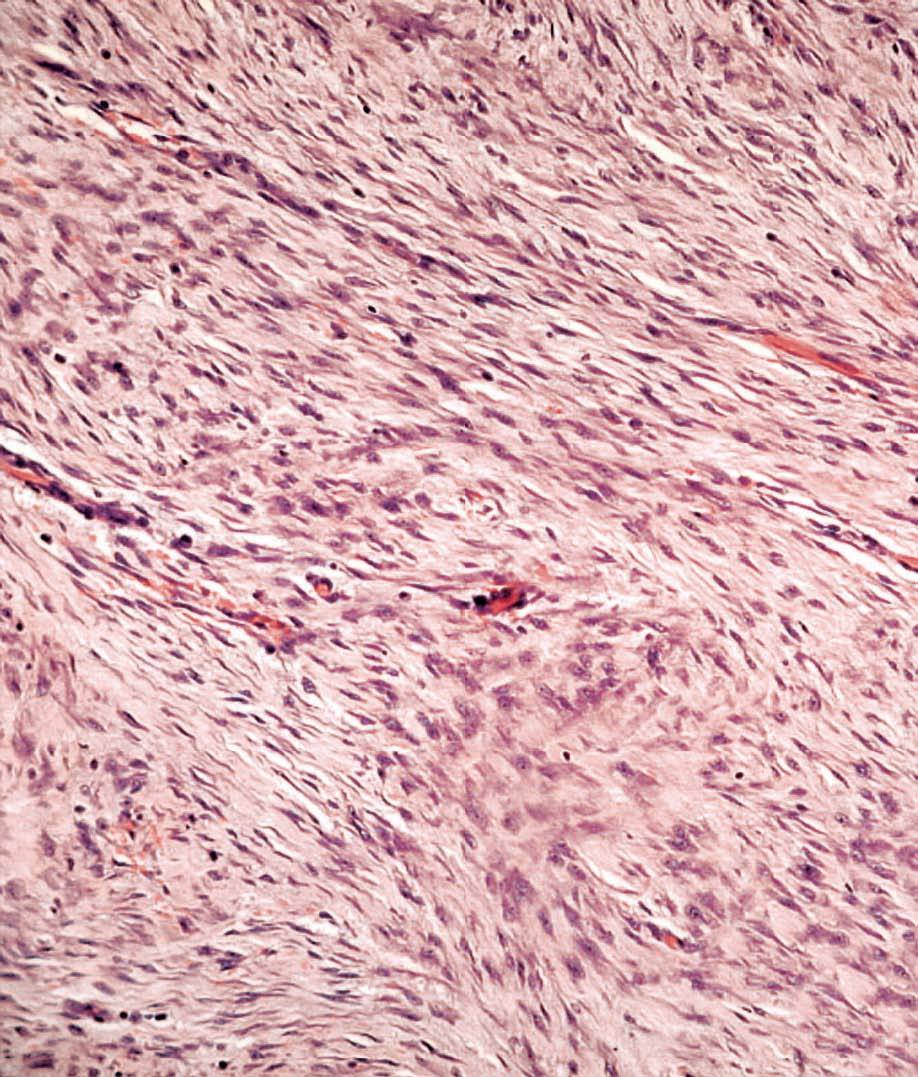
Fig. 3. Fibromatosis mamaria. A: imagen a bajo aumento en la que se aprecia la distribución en fascículos arremolinados de la tumoración. En el margen inferior se observa cómo rodean y engloban lobulillos mamarios. B: los haces entrecruzados de células fusiformes están recorridos por un delicado plexo vascular capilar.
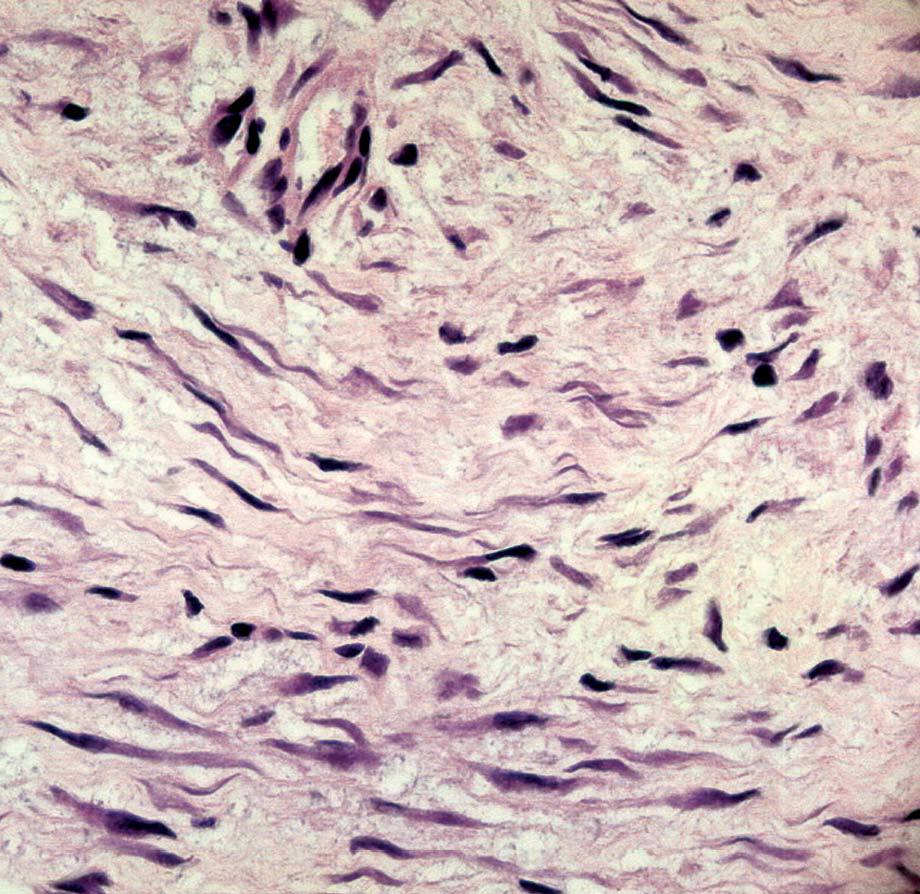
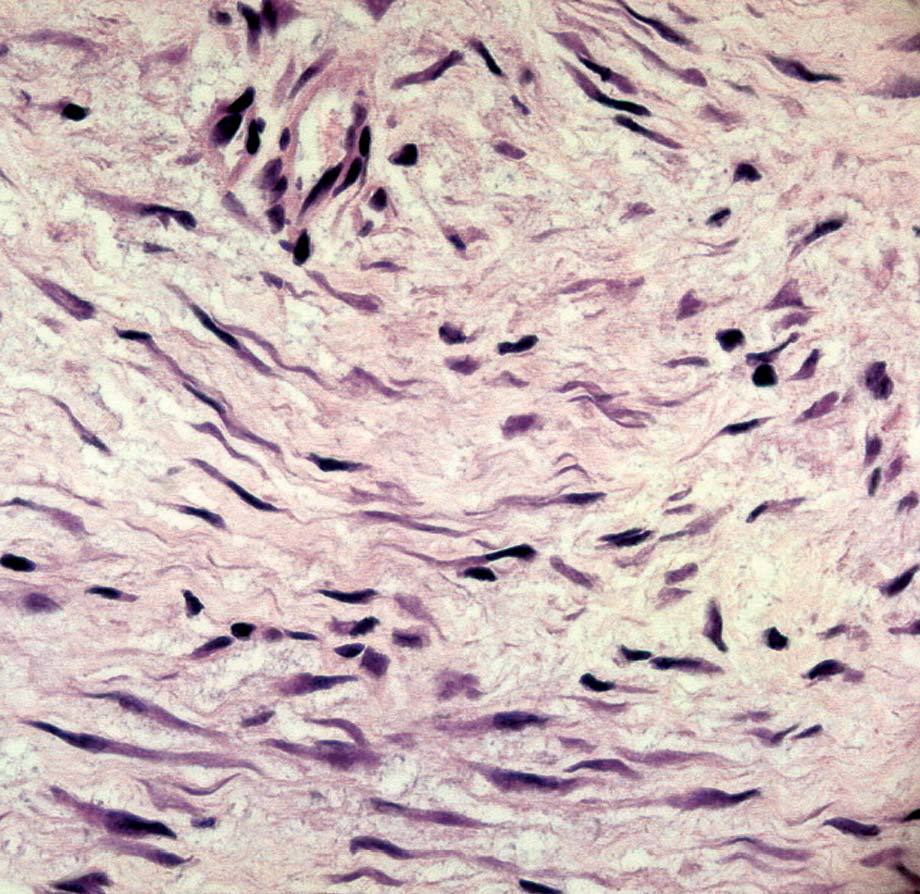
Fig. 4. Fibromatosis mamaria. A: a gran aumento se observa que citológicamente el tumor está formado por células fusiformes regulares, de núcleo alargado, cromatina densa con bajo índice mitótico. B: tinción IHQ para vimentina que muestra las células tumorales fuertemente reactivas.
La paciente no recibió tratamiento complementario y a los 9 meses del diagnóstico permanece asintomática y libre de recidiva.
DISCUSIÓN
Los tumores desmoides (fibromatosis) son tumores benignos de origen mesenquimatoso que habitualmente tienen su origen en estructuras musculoaponeuróticas profundas. El primer caso, descrito en 1832 por McFarlane, se originaba en la pared abdominal anterior1,2. El primer caso extraabdominal no fue descrito hasta 1923 por Nichols3.
Son poco frecuentes, de hecho, constituyen menos del 0,2% de los tumores primarios de mama, independientemente del sexo4.
El 49% se desarrolla en la pared abdominal, el 43% de los casos son extraabdominales y el 8% se localiza en el mesenterio. La localización más frecuente es la cintura escapular y en el músculo recto del abdomen1. La localización extraabdominal más frecuente es la pared abdominal, la cintura escapular, la región inguinal y el cuello5. En una reciente revisión de 203 casos, únicamente 9 (4%) se presentaron en la mama6.
Aunque también se han descrito casos en el varón, son más frecuentes en mujeres y sobre todo en mujeres en edad reproductiva7,8.
La etiología de la fibromatosis mamaria es desconocida, aunque se cree que es similar a las de la fibromatosis abdominal y extraabdominal. Se ha asociado con traumatismos locales y cirugía previa, como cirugía reconstructora o mamoplastia de aumento9,10 e implantes de silicona11,12. En el caso que nos ocupa, la paciente fue sometida a una tumorectomía mamaria ipsolateral 2 años antes.
Existen enfermedades genéticas con predisposición a la fibromatosis, como son el síndrome de Gardner, definido por la asociación de poliposis rectocólica familiar, tumores desmoides, osteomas múltiples en huesos faciales y quistes epidermoides13,14, fibromatosis multicéntrica familiar15 y poliposis familiar adenomatosa16,17.
Ocasionalmente, se ha asociado a embarazo y a administración de estrógenos. Tanto estos hechos como que puedan presentar receptores estrogénicos y la ocasional respuesta a antiestrógenos han hecho que se los relacione con un efecto promotor por parte de los esteroides sexuales18.
Clínicamente, en la mama se presenta como una tumoración dura, no dolorosa, mal delimitada, unilateral, de crecimiento rápido y tamaño variable, generalmente localizada y adherida a planos profundos. Puede producir retracción cutánea y de pezón, lo que hace que clínicamente parezca un carcinoma.
Radiológicamente, se incluye en el grupo de lesiones mal definidas y sospechosas de malignidad. La mamografía muestra una lesión mal definida, espiculada, sospechosa de malignidad y con ausencia de microcalcificaciones, lo que es un hallazgo característico. La ecografía muestra una masa sóli-da de márgenes irregulares, hipoecogénica, con atenuación sónica posterior con sospecha de malignidad19.
La PAAF muestra células epiteliales de aspecto atípico. La presencia de células fusiformes asociadas a pequeños grupos de células canaliculares benignas y linfocitos hace sospechar el diagnóstico20.
Su diagnóstico es histológico y se fundamenta en lesiones constituidas por fibroblastos dispuestos en haces o fascículos entrecruzados de celularidad variable, sin respuesta inflamatoria ni criterios de infiltración en el tejido circundante. El estudio microscópico muestra una proliferación de células fibroblásticas sin atipias ni mitosis, o pocas, que se organizan en fascículos largos o haces que rodean los lobulillos y los conductos lobulillares, sin respuesta inflamatoria y sin criterios de malignidad. La realización de estudios inmunohistoquímicos no es necesaria, aunque éstos muestran una positividad citoplasmática de las células fibroblásticas para la vimentina, focal para la actina y una negatividad para marcadores epiteliales, como citoqueratinas. Las estructuras glandulares se encuentran preservadas20,21.
Se debe realizar el diagnóstico diferencial con otras lesiones malignas de células fusiformes, como el carcinoma metaplásico y el fibrosarcoma, con el mioepitelioma maligno y con los tumores filoides. El diagnóstico es difícil, e incluso después de una biopsia se puede confundirla con un fibrosarcoma de bajo grado22.
Se caracterizan por un crecimiento infiltrativo local en las estructuras adyacentes, con una gran propensión a las recidivas locales, pero sin tendencia a metastatizar en los ganglios regionales o a distancia, por lo que la evaluación de enfermedad a distancia no es necesaria17.
El tratamiento adecuado es la exéresis quirúrgica del tumor con márgenes amplios para obtener un adecuado control local. En la mama el tratamiento se realiza mediante una tumorectomía amplia o una mastectomía con la finalidad de obtener bordes libres. No es necesario realizar vaciamiento axilar. La mastectomía está indicada en tumores grandes, en caso de recidiva o en casos de dificultad diagnóstica histológica21.
En el tratamiento han mostrado utilidad varios agentes farmacológicos, incluida la manipulación hormonal con tamoxifeno5 o inhibidores de la aromatasa23 y antiinflamatorios no esteroideos, interferones y agentes quimioterápicos citotóxicos24,25. El agente a emplear, la dosis y la duración del tratamiento permanecen desconocidos26. De todas formas, el tratamiento farmacológico suele reservarse para pacientes con enfermedad en progresión o tras terapia locorregional sin éxito.
El papel de la radioterapia es discutido. Parece que pacientes con enfermedad residual microscópica y/o enfermedad recurrente deberían considerarse candidatos para el tratamiento radioterápico adyuvante en función de la localización y la extensión del tumor1,27,28. Para que sea eficaz, se necesita una dosis del orden de 50 Gy. Hay autores que consideran que la radioterapia puede provocar la transformación ma-ligna de fibromatosis a fibrosarcoma, por lo que se suele reservar para recidivas o tumores irresecables17,28.
Hay autores que aconsejan la realización de un estudio colonoscópico ante el diagnóstico de fibromatosis, con la finalidad de descartar pólipos múltiples, en el contexto de un síndrome de Gardner14.
Es un tumor con alta tendencia a la recidiva local. Se estima en torno al 21%, pero inferior a la tasa de los desmoides extramamarios, que alcanza un 57%. El riesgo de recidiva depende de las características biológicas de cada tumor y generalmente aparece en los primeros 3 años tras la cirugía4,29.
En casos de tumores desmoides extraabdominales, la supervivencia global alcanza el 90% a los 10 años6.
