




La linfangioleiomiomatosis es una enfermedad pulmonar quística rara y progresiva que ocurre casi exclusivamente en las mujeres, generalmente entre la menarquia y la menopausia. Se presenta el caso de paciente de 33 años, III gesta II para, con embarazo de 32 semanas, que consultó por tos no productiva y dificultad para respirar progresiva. El examen físico fue normal, con excepción de frecuencia respiratoria elevada. La ecografía reveló un feto vivo, viable, correspondiente a 31 semanas de gestación. A la paciente se le realiza tomografía axial computarizada de alta resolución, la cual revela la presencia de cambios enfisematosos y quísticos en ambas bases pulmonares, con líquido en los bronquios. El informe de anatomía patológica de la biopsia bronquial describió engrosamiento fibromuscular difuso y pronunciado, que involucraba a los linfáticos en la capa submesotelial con proliferación de células musculares lisas y fibroblastos.
Lymphangioleiomyomatosis is a rare progressive cystic lung disease that occurs almost exclusively in women, generally between menarche and menopause. We report the case of a 33-year-old patient (gravida III, para II) with a 32-week pregnancy who presented with non-productive cough and progressive dyspnea. The physical examination was normal except for high respiratory frequency. Ultrasound examination revealed a viable live fetus corresponding to 31 weeks. High-resolution computed tomography revealed the presence of emphysematous and cystic changes in both basal lung segments and fluid in the bronchi. The bronchial biopsy report described diffuse and pronounced fibromuscular thickening and a sub-mesothelial layer with proliferation of smooth muscle cells and fibroblasts.
La linfangioleiomiomatosis (LLM) es una enfermedad pulmonar quística rara y progresiva que ocurre casi exclusivamente en las mujeres, generalmente entre la menarquia y la menopausia1. Fue reportada por primera vez por Lutembacher en 1918 en una paciente con esclerosis tuberosa, un desorden neurocutáneo autosómico dominante2. Los hallazgos claves de la LLM son la infiltración difusa del parénquima pulmonar con células musculares lisas atípicas, obstrucción del flujo de aire, neumotórax o quilotórax e insuficiencia respiratoria progresiva. Generalmente es mal diagnosticada como asma o enfermedad pulmonar obstructiva crónica3.
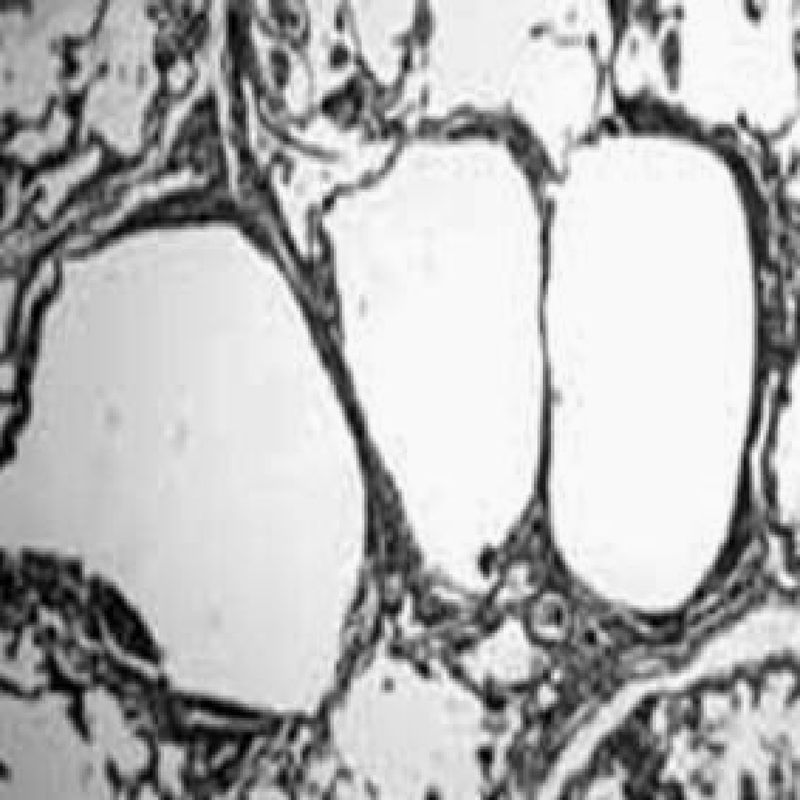
El presente caso se refiere a una paciente con embarazo de 32 semanas a la que se diagnosticó LLM (fig. 1).
Reporte del caso
Se trata de paciente de 33 años, III gesta II para, con embarazo de 32 semanas que consultó por tos no productiva y dificultad para respirar progresiva en los dos meses previos a asistir a la consulta. La paciente tenía antecedentes de asma en la infancia, pero no había utilizado ningún medicamento para esta desde los 13 años; de neumonía y de neumotórax espontáneo tres años antes del evento actual.
El examen físico fue normal, con excepción de frecuencia respiratoria de 25 rpm y una ligera disminución de los ruidos respiratorios en ambas bases pulmonares. La ecografía reveló la presencia de un feto vivo, viable, correspondiente a 31 semanas de gestación. Además, se observaron imágenes hepáticas sugestivas de fibrosis generalizada. No se encontraron alteraciones renales, ganglios retroperitoneales o ascitis.
La radiografía de tórax demostró la presencia de opacidades bronquiales lineales. La paciente es remitida al servicio de neumonología, en el cual se le indica la realización de una tomografía axial computarizada de alta resolución, la cual revela la presencia de cambios enfisematosos y quísticos en ambas bases pulmonares con presencia de líquido en los bronquios; por lo cual se realiza broncoscopio y biopsia transbronquial.
El informe de anatomía patológica describió la presencia de engrosamiento fibromuscular difuso y pronunciado, que involucraba a los linfáticos en la capa submesotelial con proliferación de células musculares lisas y fibroblastos. Al aplicar pruebas inmunohistoquímicas especiales (desmina, actina de músculo liso y vicentina), se confirmó el diagnóstico de LLM.
La paciente fue hospitalizada y tratada con broncodilatadores y oxígeno nasal, pero durante el vigésimo día de hospitalización se observó agravamiento de las condiciones clínicas y del patrón de gases arteriales, por lo cual se decide realizar cesárea obteniendo recién nacido femenino vivo de 2.150g. La paciente fue trasladada después de la cirugía a la Unidad de Cuidados Intensivos, donde muere al quinto día.
DiscusiónLa LLM ocurre esporádicamente sin evidencia genética. Es relativamente poco común con una prevalencia estimada de 2,6 casos por cada millón de mujeres. La otra forma está asociada al complejo de la esclerosis tuberosa. Ambas formas están causadas por la mutación de los genes supresores de tumores: el gen hamartin (TSC1), en el cromosoma 9 (9q34) y el locus del gen tuberina (TSC2), en el cromosoma 16 (19pI3.3)4,5. No existen factores de riesgo epidemiológicos específicos, incluyendo antecedentes familiares5.
La enfermedad ha sido descrita como la gran «imitadora», ya que las mujeres afectadas generalmente son diagnosticadas con asma, enfisema o fibrosis pulmonar. La sospecha clínica y la tomografía computarizada de alta resolución puede llevar a un diagnóstico correcto en el 80% de los casos y la biopsia puede no ser necesaria para hacer el diagnóstico definitivo. Algunas enfermedades poco comunes que presentan quistes pulmonares son: el síndrome de Birt-Hogg-Dubé, bronquiolitis folicular, neumonitis intersticial linfocítica, displasia broncopulmonar, histiocitosis de las células de Langerhans y síndrome de Sjögren3.
La pérdida de la función pulmonar se debe a la infiltración difusa del intersticio pulmonar por células musculares lisas atípicas (células linfangioleiomiomatosas), lo cual lleva a destrucción quística del parénquima pulmonar6. Se han descrito dos tipos de células características: pequeñas células de formas ahusadas y grandes células epitelioides con abundante citoplasma1. El origen de las células musculares lisas es desconocido. La recurrencia de esta condición en los pulmones trasplantados sugiere que las células provienen de un sitio remoto5. Los tumores renales benignos (angiomiolipoma) ocurren en cerca del 60% de los casos1,6. En la paciente de este caso no se observó ninguna tumoración renal.
La severidad histológica de la LLM (puntaje histológico de la LLM [PH-LLM]) puede ser cuantificado por la extensión del reemplazo del tejido pulmonar normal por las lesiones quísticas y las células linfangioleiomiomatosas. El porcentaje total de la afección del tejido por las lesiones quísticas se clasifica como sigue: PH-LLM-1=<25%, PH-LLM-2=25–50% y PH-LLM-3 >50%. La determinación del PH-LLM tiene importantes implicaciones diagnósticas, debido a que se ha observado significativa diferencia en la supervivencia entre los grupos7.
La LLM generalmente se presenta con síntomas de disnea, aparición súbita de neumotórax, derrame pleural o hemorragia intraabdominal. La disnea, la tos y la hemoptisis son los síntomas más comunes. La disnea ocurre en más del 70% de los pacientes. El tiempo promedio entre la aparición de los síntomas y el diagnóstico definitivo es de 5–6 años3. Al igual que la paciente de este informe, más del 50% de las pacientes tienen antecedentes de neumotórax. El examen físico de los pacientes con LLM es sorprendentemente normal. Sin embargo, se puede encontrar disminución de los ruidos respiratorios en pacientes con enfermedad severa y derrame pleural1.
El principal hallazgo de imágenes de la tomografía axial computarizada en las pacientes es la presencia de quistes bien circunscritos, redondos u ovalados, con paredes finas y dispersos en el pulmón. Los quistes varían en tamaño (desde unos milímetros hasta varios centímetros) y en número (desde unos pocos y escasos quistes hasta el reemplazo completo del pulmón por los quistes)1,3.
Las pruebas de función pulmonar pueden ser normales en pacientes con enfermedad temprana, aunque la mayoría tienen obstrucción del flujo y, en un tercio de los casos, se observa una ligera mejoría después de la administración de broncodilatadores. La capacidad pulmonar total está conservada, pero la transferencia de gases está marcadamente disminuida3.
No existe tratamiento que revierta efectivamente las anomalías funcionales o detenga la progresión del daño pulmonar. El tratamiento es de soporte y, generalmente, evita cualquier fármaco que contenga estrógenos, ya que se presume que este juega un papel en la alta prevalencia de la enfermedad en las mujeres. Otras medidas adicionales de soporte incluyen: terapia con broncodilatadores (20% de los pacientes tienen respuesta positiva); tratamientos para la ansiedad y la depresión; pleurodesis (en caso de neumotórax); rehabilitación pulmonar; tratamiento con suplementación de oxígeno y trasplante pulmonar. También puede ser necesario la embolización o cauterización de los tumores renales3,5. Otros tratamientos prometedores que están en investigación son la rapamicina y los inhibidores de las metaloproteinasas de la matriz y de la angiogénesis3.
Se les debe recomendar a las mujeres con LLM no embarazarse, debido a que se considera que un número significativo de pacientes desarrollarán sus síntomas iniciales durante el embarazo y que esta condición puede agravarse durante el mismo, particularmente con neumotórax y quilotórax1. No obstante, existe un informe de un embarazo exitoso en una paciente con la enfermedad8. Algunas células linfangiomiomatosas presentan receptores de estrógenos y progesterona, demostrados por estudios bioquímicos e inmunohistoquímicos. Estos hallazgos han sido el corolario biológico para la respuesta de la LLM al tratamiento hormonal9. Sin embargo, pacientes tratados con antiestrógenos no han mostrado resultados beneficiosos7. No obstante, se han reportado efectos beneficiosos con el uso de hormona liberadora de gonadotropina3.
Cuando la función pulmonar de los pacientes disminuye, el trasplante pulmonar es la mejor opción terapéutica. El trasplante pulmonar, principalmente de un pulmón, se ha realizado de forma exitosa. Las tasas de supervivencia después del trasplante pulmonar son comparables a las de pacientes con otras condiciones. La morbilidad es común después del trasplante, debido a adherencias pleurales extensas que llevan a hemorragia intraoperatoria, neumotórax del pulmón no trasplantado, quilotórax, ascitis quilosa, complicaciones de los angiolipomas renales y recurrencia del LLM en el pulmón trasplantado1,3.
