




La mastitis granulomatosa idiopática es una rara enfermedad que afecta sobre todo a mujeres jóvenes. Supone un importante desafío diagnóstico y terapéutico al presentarse en ocasiones como una lesión mamaria indicativa de malignidad. No se conoce su etiología ni existen pruebas de imagen que permitan diagnosticar con certeza la enfermedad, requiriendo de un estudio anatomopatológico para confirmar el diagnóstico. El tratamiento puede ir desde la mera observación, pasando por la toma prolongada de corticosteroides orales, hasta la cirugía. Dado que esta dolencia es infrecuente en la práctica clínica, no existen protocolos unificados acerca del mejor tratamiento y seguimiento de las pacientes. Presentamos un caso de mastitis granulomatosa idiopática diagnosticado en nuestro centro, así como el tratamiento propuesto y el seguimiento realizado. Por último, revisamos la literatura existente en relación con esta infrecuente entidad.
Granulomatous mastitis is an uncommon disease that primarily affects young women. It represents a diagnostic and therapeutic challenge, as it might be confused with breast carcinoma. There are no mammographic or ultrasonographic findings that correlate imaging features with histological results. Therapeutic management of this disease can range from simple observation to long-term corticosteroid treatment or surgery. Because granulomatous mastitis is infrequent in clinical practice, there are no standardised protocols regarding the best treatment and follow-up for these patients. We present the case of a women diagnosed in our department, and her treatment and follow-up. We also selectively examine the available literature to support our case.
La mastitis granulomatosa crónica idiopática, también denominada mastitis lobar o lobulitis granulomatosa, es una rara enfermedad inflamatoria de carácter benigno que compromete la mama, descrita por primera vez en 1972 por Kessler y Wolloch1.
Afecta sobre todo a mujeres en la tercera década de la vida2–5, más frecuentemente de etnia latinoamericana. Su origen es desconocido, aunque se ha asociado a frecuentes alteraciones inmunológicas.
Su presentación clínica más frecuente es la de una tumoración indurada y fija, de consistencia firme, más o menos dolorosa1,2,6,7, preferentemente de localización periférica respecto a la glándula mamaria, a diferencia de la mastitis periductal, de presentación más central; ocasionalmente se asocia a eritema y ganglios palpables8. En la mayoría de las ocasiones se presenta de forma unilateral, siendo bilateral en un 1-9% de ellas5,7.
Resulta primordial para el diagnóstico la confirmación anatomopatológica de la muestra del tejido afecto, así como descartar la asociación de enfermedades autoinmunes (diabetes, disfunción tiroidea, sarcoidosis o enfermedad relacionada con IgG4)9.
El diagnóstico de esta entidad debe ser de exclusión, basándose en la confirmación histológica. Del mismo modo, es imprescindible descartar malignidad y otras causas de enfermedad granulomatosa1,2,6,7.
El tratamiento no ha sido aún establecido, con medidas que van desde la observación y la corticoterapia, a la cirugía (incluida la mastectomía)6–10.
A pesar de ser una enfermedad en la que pueden existir recurrencias3,6, el pronóstico es favorable, pudiendo resolverse el cuadro en menos de un año en la mayoría de los casos2.
A continuación exponemos el caso clínico de una paciente con mastitis granulomatosa, el manejo realizado y revisamos la literatura publicada al respecto.
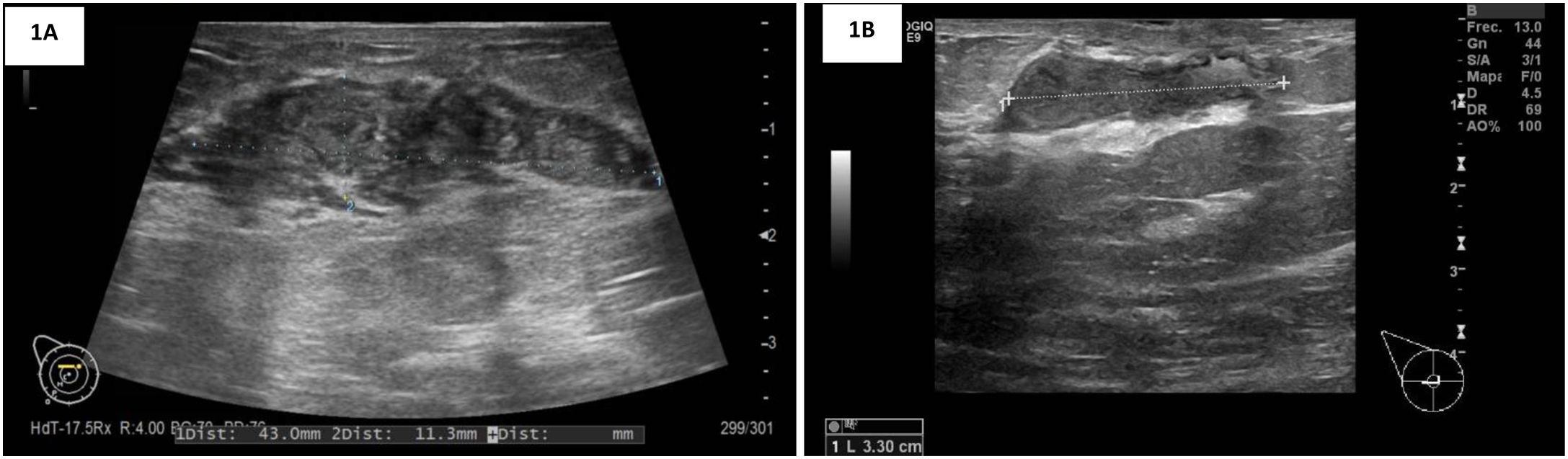
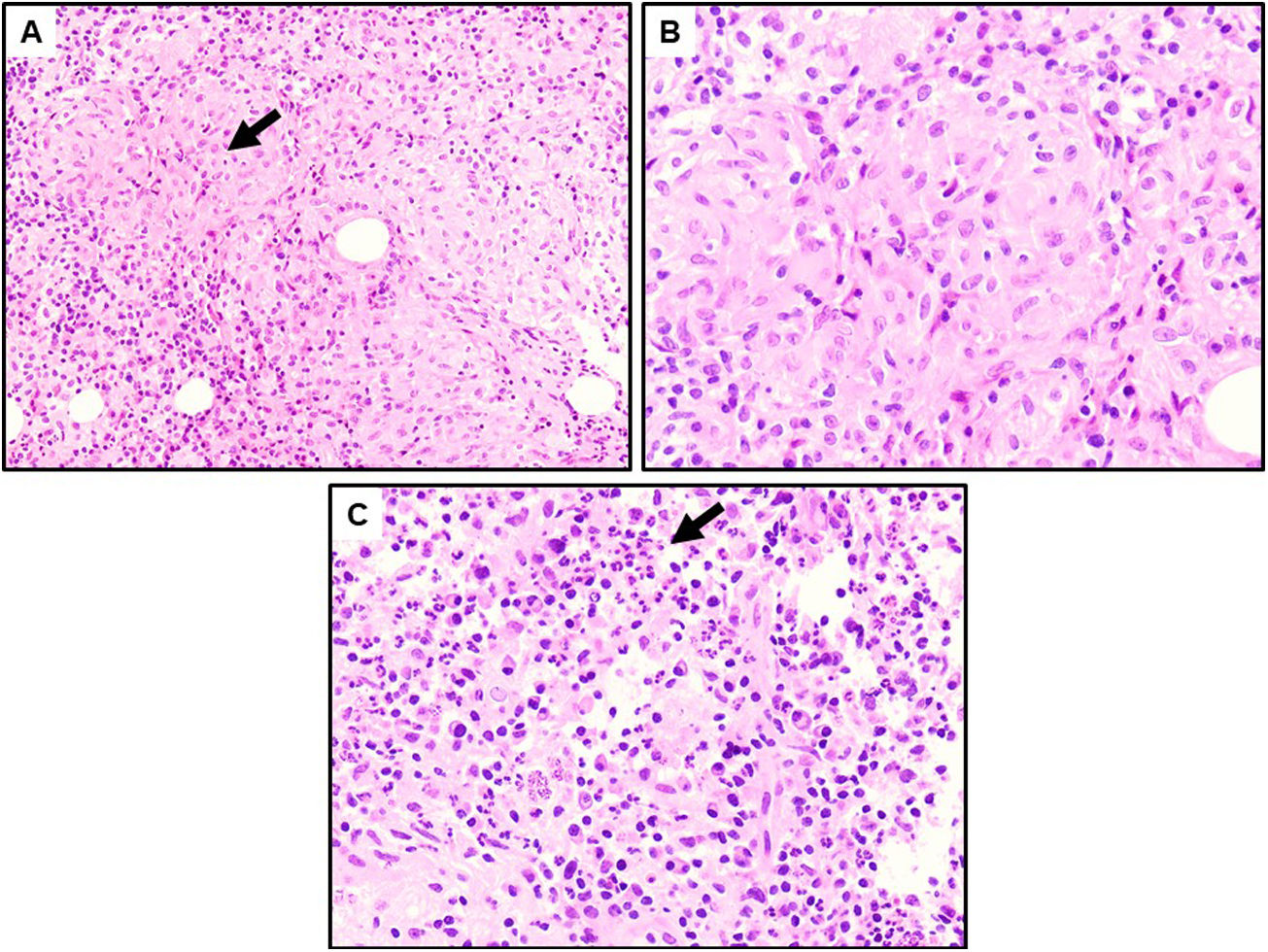
Caso clínicoPaciente mujer de 32 años, originaria de Sudamérica (Colombia), que consultó en Urgencias por autopalpación de nódulo mamario, sin antecedentes personales o familiares de interés. La exploración mamaria evidenció un nódulo retroareolar hacia cuadrantes externos de la mama derecha de unos 40mm. En la ecografía mamaria se observaba una extensa área de unos 50mm de longitud×15mm de espesor asociada a una lesión nodular polilobulada, hipoecogénica y heterogénea, con márgenes irregulares (fig. 1A). Se catalogaron las lesiones como BI-RADS 4C. A nivel axilar se identificaron al menos 2 ganglios con cortical engrosada, Bedi 2. Tras la realización de una biopsia con aguja gruesa del tejido mamario implicado y una punción-aspiración con aguja fina axilar, el estudio anatomopatológico confirmó una mastitis granulomatosa (fig. 2).
: área de 43×11mm asociada a una lesión nodular polilobulada, hipoecogénica y heterogénea, con márgenes irregulares. BI-RADS 4C. B: Ecografía mama derecha CSE: área hipoecogénica de unos 33mm. BI-RADS 2.")
 rodeados por un proceso inflamatorio de predominio linfoplasmocitario (HE, ×10). B: Población de macrófagos agregados que forman granulomas no caseificantes rodeados de células inflamatorias (HE, ×20). C: Abundante población de neutrófilos en un fondo de linfocitos y células plasmáticas, frecuentemente asociados a mastitis granulomatosa idiopática (HE, ×20).")
A: Granulomas no necrosantes (flecha) rodeados por un proceso inflamatorio de predominio linfoplasmocitario (HE, ×10). B: Población de macrófagos agregados que forman granulomas no caseificantes rodeados de células inflamatorias (HE, ×20). C: Abundante población de neutrófilos en un fondo de linfocitos y células plasmáticas, frecuentemente asociados a mastitis granulomatosa idiopática (HE, ×20).
El estudio de la paciente se completó con la realización de pruebas analíticas que descartaron diabetes mellitus (HbA1c 5,9%) y enfermedades autoinmunes. Las pruebas serológicas (virus de la inmunodeficiencia humana, virus de las hepatitis B y C), así como el estudio microbiológico, descartaron un origen infeccioso; además, la tinción ácido-alcohol resistente excluyó la tuberculosis como causa de granuloma.
Una vez alcanzado el diagnóstico, se consensuó con la paciente inicialmente mantener una actitud expectante sin tratamiento; sin embargo, se produjo un aumento de la sintomatología dolorosa y la extensión de la lesión mamaria, que propiciaron el inicio de corticoterapia oral (prednisona 50mg/día), objetivándose una reducción clínica de la lesión palpable ya evidente tras la primera semana de tratamiento.
La ecografía realizada 3 meses más tarde identificó un área hipoecogénica de unos 33mm de eje máximo, comprobándose una disminución significativa del tamaño de la lesión previamente existente, catalogada como BI-RADS 2 (fig. 1B). Clínicamente era prácticamente indetectable.
Ante la mejoría clínica se decidió mantener la misma pauta de corticoides durante 4 meses (presentando efectos secundarios leves como insomnio y edema facial que no precisaron tratamiento adicional), y posteriormente se redujo progresivamente la dosis en 5mg cada 2 semanas hasta completar 9 meses de tratamiento.
Transcurridos 2 meses sin tratamiento, y en el contexto de una gestación incipiente, la paciente presentó un nuevo episodio de mastitis aguda. Se administró antibiótico oral (ciprofloxacino 500mg/12h durante 14 días) y se produjo un drenaje espontáneo del material purulento, sin precisar de intervención quirúrgica. La ecografía mamaria realizada una vez resuelto el cuadro de mastitis aguda no objetivó nódulos ni lesiones mamarias previamente existentes, por lo que se dio por resuelto el cuadro de mastitis granulomatosa previo.
Tanto la evolución como la finalización del embarazo transcurrieron sin complicaciones. Se realizó una cesárea programada por 2 cesáreas anteriores, sin incidencias. Fue durante el puerperio, y tras iniciar la lactancia materna, cuando tuvo que acudir a Urgencias por un nuevo episodio de mastitis en la mama previamente afecta, que se resolvió con tratamiento antibiótico oral (cefadroxilo 500mg/12h durante 7 días).
Actualmente la paciente mantiene la lactancia materna, por lo que se ha consensuado demorar el seguimiento clínico en la Unidad de Mama hasta que finalice este periodo.
DiscusiónLa incidencia de la mastitis granulomatosa es desconocida, en torno a 1,8% según algunas series6, siendo más frecuente en mujeres latinas y asiáticas6.
El origen de esta entidad es desconocido, si bien se relaciona con un daño epitelial ductal, que provoca la transición de las secreciones luminales ricas en proteína y grasa hacia el tejido conectivo lobar, provocando una inflamación local con la consiguiente migración de macrófagos y linfocitos a la región, que provoca una respuesta inflamatoria granulomatosa local7,8. En cuanto a la etiología del factor que desencadena el daño endotelial, la más aceptada es la teoría inmunológica, reforzada por la respuesta al tratamiento con inmunosupresores. Aunque las pruebas serológicas clásicas de trastornos autoinmunes, como factor reumatoide o anticuerpos antinucleares, suelen ser negativas4,8 deben descartarse enfermedades autoinmunes como la diabetes, la disfunción tiroidea, la sarcoidosis o la enfermedad relacionada con IgG49. Los niveles elevados de prolactina se han descrito como un posible desencadenante, por su papel en el aumento de las secreciones ductales, lo que daña el epitelio ductal7,11.
No se ha definido un patrón característico que permita identificar la mastitis granulomatosa idiopática en las pruebas de imagen. La mamografía presenta un escaso valor predictivo positivo para diferenciarla de los carcinomas mamarios, al mostrar densidades focales asimétricas con márgenes irregulares5,8,10. En el caso descrito se apostó por el diagnóstico mediante ecografía, que aunque presenta baja especificidad, permite además guiar la biopsia que nos dará el diagnóstico definitivo. Los hallazgos descritos en las pruebas de imagen se muestran según la literatura como lesiones predominantemente hipoecoicas con sombra y refuerzo acústico posterior, a menudo agrupadas con extensión tubular a modo de tentáculos, que se insinúan alrededor de los lobulillos mamarios5,7,8. Estos hallazgos coinciden con los encontrados en nuestra paciente. El estudio ecográfico se puede completar con la valoración Doppler, que mostrará un aumento inespecífico de la vascularización7. Estudios recientes apuestan por el uso de las imágenes de resonancia magnética, ya que permiten una delimitación precisa de la inflamación, de su extensión y del control evolutivo de la enfermedad5,8,11. Esta entidad se manifiesta en la resonancia magnética con un patrón heterogéneo y mal definido con realce no masa con cinética de realce periférico tras la administración de gadolinio7,8. Otros hallazgos adicionales que pueden visualizarse incluyen: la linfadenopatía axilar, el engrosamiento del pezón y/o la piel, la presencia de tractos sinusoidales y la distorsión del parénquima7. El análisis del coeficiente de difusión aparente puede servir como una herramienta para diferenciar la mastitis granulomatosa idiopática del carcinoma en aquellas lesiones no masa con borde realzado5. Aunque pueden existir falsos positivos para malignidad, las curvas de intensidad de tiempo son más benignas, más consistentes con la inflamación, con un patrón de difusión menos restringido que para el carcinoma7,11.
Sin embargo, el diagnóstico definitivo será confirmado mediante estudio anatomopatológico de la muestra extraída, permitiendo así descartar malignidad del proceso. Los hallazgos encontrados revelarán una lobulitis granulomatosa crónica no caseificante4,6–8,11,12. Puede además observarse células multinucleadas tipo Langerhans, linfocitos y microabscesos7. Es importante realizar estudios microbiológicos (incluyendo micobacterias, hongos y materiales extraños) que permitan descartar todas las causas que puedan generar una reacción granulomatosa, como realizamos en nuestra paciente.
Dada la baja frecuencia del diagnóstico, no disponemos actualmente de guías clínicas y la mayoría de la información se obtiene a partir de las series de casos. Algunos grupos, como el de Davis, afirman que se trata de un proceso autolimitado que se resuelve aproximadamente en un periodo de 5 meses2, sin encontrar diferencias significativas con las terapias activas. Este manejo está indicado sobre todo para los casos recientes con síntomas leves a moderados. A menudo las pacientes son sometidas a ciclos de antibiótico, ya que puede simular una mastitis piógena, sin mejoría y con un aumento del tiempo de resolución de la misma2. Se ha encontrado asociación con el Corynebacterium8,11, bacilo de difícil cultivo que a menudo forma parte de la flora normal de la piel. En estos casos, a menudo se presenta con fiebre, neutrofilia y presentación bilateral, respondiendo al tratamiento con clindamicina8. No obstante, es necesario recordar que el diagnóstico requiere de la exclusión de otras casusas de granulomas.
En cuanto al tratamiento activo, debemos distinguir diferentes escenarios. Para la enfermedad clínicamente avanzada o síntomas más graves, se recomienda el uso de corticoterapia, con ventajas en la disminución y remisión de la enfermedad. La mayor experiencia se encuentra con prednisona por vía oral, con dosis iniciales de 0,8mg/kg/día13 o 30-60mg/día8, disminuyendo progresivamente la dosis con la mejoría clínica. La mayor limitación para esta terapia se encuentra en los efectos secundarios de este tipo de tratamientos. La administración local pretende solventar los mismos. Se ha descrito su uso por vía tópica y mediante inyección intramamaria guiada por ultrasonidos, con dosis de 40mg/ml de acetato de metilprednisolona. El grupo de Alper consigue una respuesta completa en el 32% de las pacientes con inyecciones repetidas entre 2-7 veces4. Para casos refractarios se han empleado agentes como el metotrexato, con respuesta variable y escasa evidencia clínica, observándose tasas de recaída del 17,6%14. Se han descrito tratamientos con azatioprina, hidroxicloroquina o colchicina13,14, empleados en la mayoría de los casos como ahorradores de corticosteroides, y no como tratamiento aislado8.
La cirugía, mediante escisión de la lesión, proporciona una mayor precisión diagnóstica6, si bien debe retrasarse hasta el control de los síntomas inflamatorios agudos para limitar la aparición de cicatrices. A tal efecto se recomienda el uso concomitante en el postoperatorio con esteroides para mejorar el resultado8.
El pronóstico de la enfermedad es favorable, con un tiempo promedio de resolución que va desde los 5 a los 12 meses2. Las recurrencias varían en torno al 5-67% según distintas series, siendo más frecuentes los primeros meses tras la resolución del primer caso3,8. Pese a su presentación en ocasiones indicativa de neoplasia, la malignización es excepcional6. Su asociación con el cáncer de mama es rara, habiéndose descrito tan solo 8 casos desde 19386.
FinanciaciónLos autores declaran no tener financiación.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
