




INTRODUCCIÓN
El síndrome de Lowe, también conocido como síndrome oculocerebrorrenal, es una enfermedad hereditaria recesiva asociada al cromosoma X, extremadamente rara, que se caracteriza clínicamente por anomalías oculares (cataratas congénitas, glaucoma), afección del sistema nervioso central (hipotonía generalizada con disminución o ausencia de los reflejos osteotendinosos y retraso mental severo), alteraciones renales (disfunción renal progresiva con acidosis e hiperaciduria) y facies peculiar1.
Fue descrito por primera vez en 1951 por Charles Lowe. En España, su incidencia, según datos del ECEMC (Estudio Colaborativo Español de Malformaciones Congénitas), se estima en 0,013 casos por 10.000 recién nacidos vivos2.
Se presenta clínicamente sólo en varones, y las mujeres portadoras son asintomáticas.
Excepcionalmente se han descrito algunas mujeres con manifestaciones clínicamente evidentes, pero de carácter leve3.
Se hereda como un rasgo genético recesivo ligado al cromosoma X, y se ha identificado el locus causal del defecto genético en la región q26 (Xq25-26,1) y el gen causante parece codificar una proteína muy semejante al inositol fosfato-5-fosfatasa localizada en el aparato de Golgi. Esto se relaciona con un déficit en la absorción intestinal de aminoácidos del tipo lisina o arginina, que conduce a un fallo en el metabolismo de proteoglucanos o glucosaminoglucanos, sustancias que participan en la síntesis del colágeno que producen los fibroblastos y que están implicadas en la integridad de los fotorreceptores2.
Clínicamente, desde los primeros meses de vida cursa con alteraciones oculares y retraso mental acusado y progresivo, miopatía y neuropatía con hipotonía e hiporreflexia graves durante el primer año. Presenta un amplio espectro clínico, en todos los casos es constante la aparición de cataratas congénitas bilaterales nucleares y densas, y son frecuentes las anomalías en la conducta4. Las cataratas frecuentemente se asocian a adelgazamiento corneal que se manifiesta por la presencia de escleróticas azules, glaucoma, malformación del ángulo de la cámara anterior y del iris, y en menor proporción se asocian a microftalmía, pupilas mióticas y enoftalmos5. Estas alteraciones, junto a la posible ausencia de cejas, les proporciona una facies peculiar.
La afección renal del túbulo proximal da lugar a una acidosis tubular renal, tipo síndrome de Fanconi, con hiperaminoaciduria generalizada y proteinuria. Las manifestaciones renales suelen comenzar con poliuria, anorexia, vómitos, acidosis metabólica y signos de raquitismo hipofosfatémico, resistente a vitami na D2.
Los pacientes con este síndrome pueden presentar osteoporosis, criptorquidia, contracturas articulares y excepcionalmente acompañarse de convulsiones, pectus excavatum, quistes dentales y cálculos renales6,7.
El síndrome de Lowe tiene un pronóstico desfavorable condicionado por la severidad del retraso psicomotor y el desarrollo de ceguera en los pacientes, que suelen fallecer por fallo renal progresivo antes de los 40 años2,5,8.
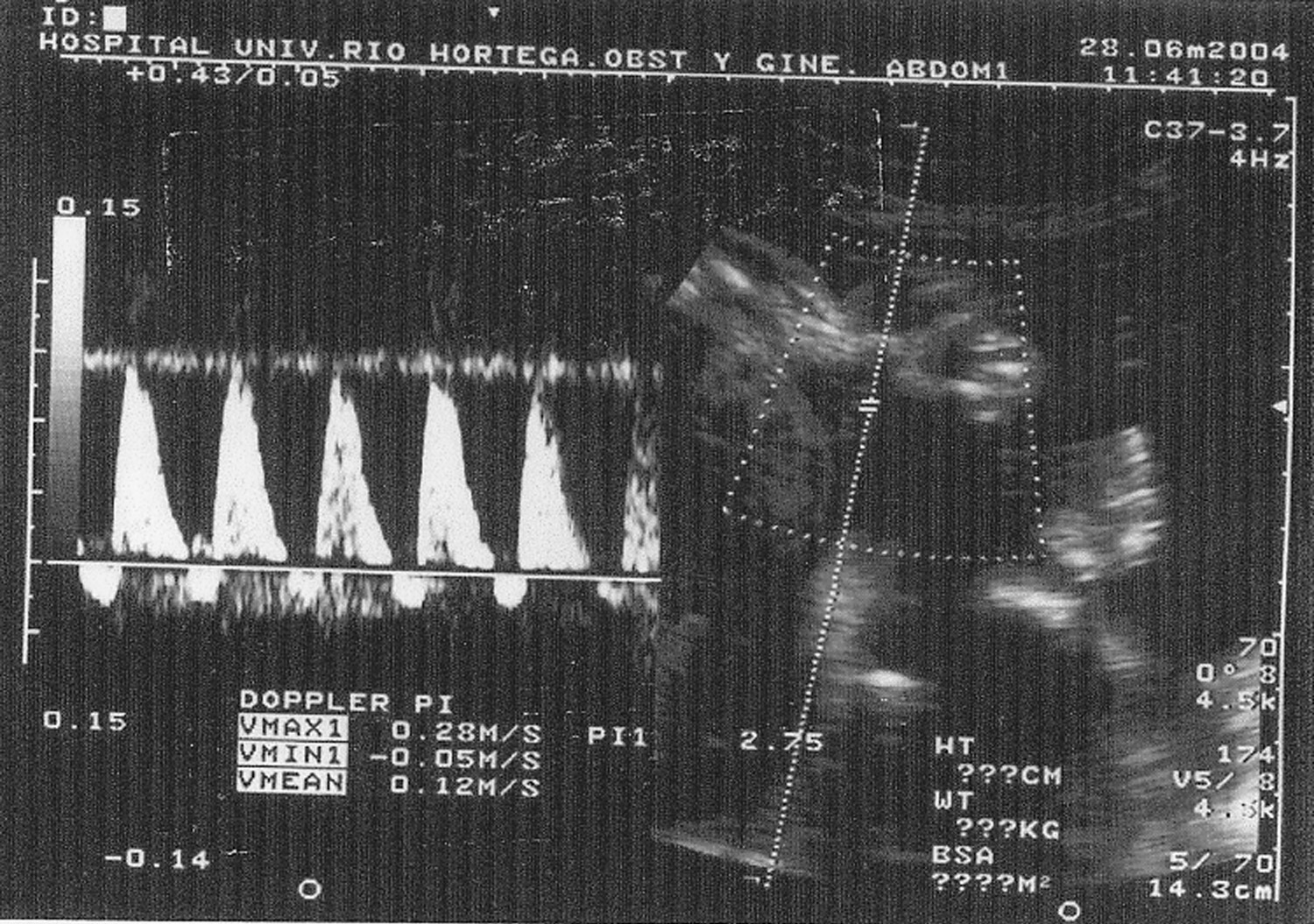
Fig. 1. Doppler en arteria umbilical con índice de resistencia elevado y flujo diastólico reverso.
Se recomienda ofrecer consejo genético ante la sospecha prenatal de síndrome de Lowe o historia familiar positiva. La sospecha prenatal se basa en el hallazgo de concentraciones elevadas de alfafetoproteína tanto en el suero materno como en el líquido amniótico, en ausencia de actividad colinesterasa, que parece relacionarse con la alteración renal fetal9,10. Esto obliga al rastreo de portadores en la familia.
No existe tratamiento curativo específico; las medidas terapéuticas deben basarse en el tratamiento de soporte de las complicaciones oculares y del síndrome de Fanconi (aporte de electrólitos, diuréticos y antiinflamatorios no esteroideos)2. El tratamiento quirúrgico para la extracción de las cataratas se asocia a un mal pronóstico en cuanto a recuperación visual completa5.
CASO CLÍNICO
Nos encontramos ante una mujer de 31 años, grupo sanguíneo A positivo, portadora asintomática del síndrome de Lowe (diagnosticada por historia familiar materna y análisis del ADN), sin otros antecedentes de interés. En su primer embarazo (marzo de 2002), se realizó una interrupción voluntaria por el diagnóstico, mediante amniocentesis y análisis del ADN, de un feto varón afectado por este síndrome.
En noviembre de 2003 se realizó una fertilización en vitro (FIV) con selección del sexo embrionario, y se consiguió una gestación gemelar bicorial biamniótica, con un gemelo muerto intraútero a las 16 semanas de gestación (enero de 2004). No consta un nuevo control ecográfico, hasta que la paciente es remitida a nuestro servicio; en la ecografía se objetiva un feto mujer con un retraso de crecimiento intrauterino severo (35 semanas de amenorrea acorde a 27 semanas), con Doppler en arteria umbilical patológico (índice de resistencia de arteria umbilical muy elevado y flujo diastólico reverso) (fig. 1), por lo cual se decide la realización de una cesárea urgente. Nace un feto vivo hembra de 720 g, Apgar 8/9 y aspecto de retraso de crecimiento intrauterino (CIR) severo, que hace preciso su traslado a incubadora.
Durante los 3 primeros días de evolución, la recién nacida presentó distrés respiratorio transitorio, hipoglucemia precoz transitoria (glucemia inicial de 19 mg/dl que se solucionó con aportes iniciales de glucosa de 7 g/kg/día), trombocitopenia leve (66.000 plaquetas), ictericia multifactorial (cifra inicial de bilirrubina de 15,6 mg/dl, con bilirrubina directa de 1,3 mg/dl); todo ello se interpretó en el contexto de CIR severo. Debido a la gravedad del retraso de crecimiento intrauterino y la enterocolitis necrotizante, se decidió el traslado de la recién nacida a un hospital con disponibilidad de cirugía pediátrica.
DISCUSIÓN
El síndrome de Lowe es una enfermedad extremadamente infrecuente (0,013 casos por 10.000 recién nacidos vivos)2, lo que convierte la detección de un caso en un tema de interés. Como ya hemos comentado, se trata de un síndrome que padecen los varones, al estar su transmisión ligada al cromosoma X. Aunque existen algunos casos descritos de mujeres con síndrome de Lowe3, en ellas las manifestaciones clínicas son leves y la mayoría son portadoras totalmente asintomáticas, como nuestra paciente. Gran parte de estas mujeres ignoran su condición de portadoras, lo que imposibilita la aplicación de métodos de diagnóstico prenatal para la enfermedad en sus descendientes. En este caso sí conocíamos el estado de portadora de la mujer, porque hay una historia familiar de consanguinidad y antecedentes de un caso de síndrome de Lowe, ya que se estudió a toda la familia. Gracias a esto, en la primera gestación se pudo aplicar el diagnóstico prenatal mediante la amniocentesis, con análisis del ADN fetal que mostró un cariotipo XY afectado de este síndrome, y se realizó una interrupción voluntaria de la gestación.
Puede hacerse en el primer trimestre un diagnóstico de sospecha más precoz, mediante el análisis en suero materno o amniótico de alfafetoproteína, cuyos valores se encuentran elevados9. Éste es un marcador muy inespecífico que se eleva en otras afecciones, como los defectos del tubo neural, obstrucciones gastrointestinales, higroma quístico, carcinoma hepatocelular materno, etc. El diagnóstico de certeza lo da el análisis cromosómico del ADN mediante la amniocentesis del segundo trimestre. Se ha logrado incluso detectar mediante hibridación in situ fluorescente (FISH) la mutación en el gen y el ARN mensajero defectuoso en las células amnióticas cultivadas10,11. En casos de historia familiar conocida, como éste, la aplicación de técnicas de diagnóstico más precoces, como la biopsia corial, también resultan factibles, aunque no debemos olvidar el mayor riesgo de pérdida embrionaria que ésta supone (1-5%) comparada con la amniocentesis (0,5-1%)12.
La paciente optó por recurrir a técnicas de reproducción asistida (FIV), que permiten la selección de los embriones a transferir una vez analizado su sexo.
No consta en la historia que la paciente se realizara amniocentesis en el curso de esta gestación gemelar. No debemos olvidar que aunque ambos fetos fueran de sexo femenino y por tanto no estuvieran afectados del síndrome de Lowe, el hecho de que la gestación fuera conseguida mediante FIV aumenta el riesgo de cromosomopatías respecto a la población general (debido a la manipulación y selección embrionaria).
No podemos afirmar que la muerte intrauterina del primer gemelo en la semana 16 ni el CIR severo del segundo fueran causados por una cromosomopatía, pero sí que hubiera sido aconsejable el estudio del cariotipo de ambos fetos mediante amniocentesis, para descartar así las cromosomopatías más frecuentes, además del estado de portador del síndrome de Lowe. Sin embargo, hay que recordar que esta decisión, con una correcta y objetiva información médica, corresponde exclusivamente a la pareja.
El pronóstico de la recién nacida, que aún vive, es sombrío, dados su bajo peso al nacer (720 g), su estado de CIR severo y las alteraciones metabólicas y analíticas acompañantes, así como el distrés respiratorio y la aparición de enterocolitis necrotizante. Lo mismo podemos decir del futuro genésico de la madre, cuyo estado de portadora del síndrome de Lowe la hace candidata a la realización de pruebas invasivas de diagnóstico prenatal e incluso preimplantacional si recurre de nuevo a técnicas de reproducción asistida.
Nos parece interesante la descripción de un síndrome tan poco frecuente y las repercusiones que tiene no sólo en los enfermos, sino en la descendencia de los portadores sanos, así como el papel importante que tienen las pruebas diagnósticas prenatales en su detección. Los avances científicos en el campo de las técnicas de reproducción asistida, como la selección embrionaria en función del sexo para evitar la aparición de las enfermedades ligadas al cromosoma X, suponen una gran ayuda y esperanza para las parejas afectadas de síndromes similares a éste, aunque en este caso el resultado de un recién nacido sano y a término (que es el objetivo de toda técnica de reproducción asistida) no ha sido posible.
