



To validate the importance of the angiotensin II receptor isotypes and the lymphatic vessels in systemic sclerosis and idiopathic pulmonary fibrosis.
METHODS:We examined angiotensin II type 1 and 2 receptors and lymphatic vessels in the pulmonary tissues obtained from open lung biopsies of 30 patients with systemic sclerosis and 28 patients with idiopathic pulmonary fibrosis. Their histologic patterns included cellular and fibrotic non-specific interstitial pneumonia for systemic sclerosis and usual interstitial pneumonia for idiopathic pulmonary fibrosis. We used immunohistochemistry and histomorphometry to evaluate the number of cells in the alveolar septae and the vessels stained by these markers. Survival curves were also used.
RESULTS:We found a significantly increased percentage of septal and vessel cells immunostained for the angiotensin type 1 and 2 receptors in the systemic sclerosis and idiopathic pulmonary fibrosis patients compared with the controls. A similar percentage of angiotensin 2 receptor positive vessel cells was observed in fibrotic non-specific interstitial pneumonia and usual interstitial pneumonia. A significantly increased percentage of lymphatic vessels was present in the usual interstitial pneumonia group compared with the non-specific interstitial pneumonia and control groups. A Cox regression analysis showed a high risk of death for the patients with usual interstitial pneumonia and a high percentage of vessel cells immunostained for the angiotensin 2 receptor in the lymphatic vessels.
CONCLUSION:We concluded that angiotensin II receptor expression in the lung parenchyma can potentially control organ remodeling and fibrosis, which suggests that strategies aimed at preventing high angiotensin 2 receptor expression may be used as potential therapeutic target in patients with pulmonary systemic sclerosis and idiopathic pulmonary fibrosis.
Diffuse parenchymal lung diseases (DPLD), also known as interstitial lung diseases (ILD), encompass a heterogeneous group of disorders, including idiopathic pulmonary fibrosis (IPF) and systemic sclerosis (SSc). These disorders result from the remodeling of the lung parenchyma by varying inflammation and fibrosis patterns that frequently affect the alveolar septae and vessels and their respective epithelial, endothelial and muscular linings (1). However, patients with interstitial pneumonia associated with SSc have a better prognosis than patients with idiopathic interstitial pneumonia (2-6) because of their increased response to immunosuppressive therapies and the prevalence of septal cellular thickening, which is associated with the pattern of nonspecific interstitial pneumonia (NSIP) (7-12). The prognosis is not as good for IPF patients with a pattern of usual interstitial pneumonia (UIP) (12). Thus, there is great interest in developing methods that can identify the mechanisms of parenchymal and vascular remodeling. We must identify these pathogenic mechanisms to avoid destroying the lungs and to provide effective treatment.
Many studies have investigated the parenchymal and vascular remodeling markers in DPLD to identify the mechanisms behind the disease progression and shortened survival rates (13-17). Because epithelial and endothelial cells and fibroblasts represent an abundant and functionally important group of cells in lung fibrosis (18), a group of angiotensins that exert biological effects on these cell types have been described (19) and, therefore, may be targeted as potentially useful modulators of organ remodeling and fibrosis (20-22). Of these angiotensins, angiotensin II (ANG II) has shown promise. ANG II is generated by the cleavage of angiotensin I by the angiotensin-converting enzyme (ACE). ANG II binds to and acts via 2 high-affinity receptor isotypes, the angiotensin II type-1 (AGTR-1) and type-2 (AGTR-2) receptors, both of which belong to the 7-transmembrane G protein–coupled receptor family (23,24). In some studies, AGTR-2 has been detected in different adult human cells and in fetal tissue, in which it seems to play a major role during embryonic development (25-27). Other studies have shown that AGTR-2 inhibits or counteracts the AGTR-1 mediated effects by directly interacting with AGTR-1 (28), but there has been uncertainty about the signaling pathway activated by AGTR-2 (29-31). These studies have indicated that the ACE-ANGII-AGTR axis contributes to the fibrotic response in the lung; however, the role that the AGTRs play in developing lung fibrosis in IPF and SSc remains unclear. To validate the importance of the ACE-ANGII-AGTR axis, to explore the quantitative relationship between this factor and patient outcomes and to examine the relationship between AGTRs and other parenchymal and vascular factors, we studied these markers in 30 SSc and 26 IPF patients.
PATIENTS AND METHODSBetween January 2002 and July 2004, 30 consecutive patients with SSc and ILD (identified using high-resolution computed tomography [HRCT]) and 26 IPF patients underwent open lung biopsies at the Clinical Hospital, University of São Paulo Medical School (1,32). The median patient age was 64 years (range, 57-71 years) for IPF (16 men, 10 women) and 45 years (range, 35-53 years) for SSc (all women). All patients fulfilled the diagnostic and subtype criteria for SSc (33,34) and IPF (35,36). Open lung biopsy was performed using formal thoracotomy, thereby avoiding honeycombing areas. The 56 patients provided written informed consent (protocol number 0960/08), and the study was approved by the local ethics committee. The patients' clinical records were analyzed. The HRCT and pulmonary function test (PFT) were performed up to 3 months before the biopsies. The pulmonary function tests included the forced expiratory volume in 1 s (FEV1), forced vital capacity (FVC), FEV1/FVC ratio 100, total lung capacity (TLC) and residual volume and carbon monoxide transfer factor (DLCO). The TLC, residual volume and residual volume/TLC percentages were measured using the helium dilution method with a MasterScreen apparatus (Erich Jaeger GmbH, Würzburg, Bavaria, Germany), and the DLCO and DLCO/alveolar volume were measured using the single breath-holding helium dilution method (37). The lung function measurements are reported as percentages of the predicted values. The cardiac parameters of these patients were normal. The HRCTs were performed using 1.0- or 1.5-mm-thick sections with patients in the supine position and with full inspiration at 10-mm intervals. A specialized chest radiologist and a pneumologist analyzed the images at 3 pre-established levels (i.e., the trachea, carina and pulmonary veins) for the presence of any signs of ILD, such as ground glass, consolidation, reticular opacities, honeycombing and bronchiectasis.
Histological analysisThe pulmonary tissue was fixed in 10% neutral buffered formalin and embedded in paraffin. Thin sections were stained with hematoxylin and eosin. Additional subserial sections from the paraffin blocks were used for the immunohistochemistry. Two pathologists who specialize in lung diseases and were blinded to the clinical aspects of the patients classified the lung specimens according to the new ILD classification consensus (1). The pathologists made their final diagnoses by consensus. UIP, the histologic pattern of IPF, was characterized by a patchy subpleural and paraseptal distribution of the parenchymal injury. Temporal heterogeneity was observed at low magnification, with areas of normal lung parenchyma alternating with alveolar collapse, interstitial mononuclear infiltrates, septal fibromyxoid tissue (fibroblastic foci) and honeycomb lung. All SSc patients had histologic patterns that were consistent with non-specific interstitial pneumonia, as defined by temporally homogeneous septal inflammatory thickening and interstitial fibrosis. Normal lung tissue, which was obtained from 10 individuals (3 males and 7 females; median age 47 years, range 31 to 60 years) who had died suddenly from non-pulmonary causes, served as the control.
ImmunohistochemistryA standard peroxidase technique was used with Harris's hematoxylin as the counterstain to identify AGTR-1 and AGTR-2 expression in the alveolar septae and vascular wall cells. D2-40 was used to identify lymphatic vessels in the control, IPF-UIP pattern (normal, collapsed and fibroblastic foci areas) and SSc-NSIP pattern lungs. Biotinylated goat polyclonal antibodies were used. Anti-AGTR-1, anti-AGTR-2 (Santa Cruz Biotechnology, Inc., Santa Cruz, CA, USA) and anti-D2-40 (Signet Laboratory Inc., USA) antibodies were incubated with the tissue sections at 1:50, 1:50 and 1:100 dilutions, respectively. A Max Polymer Novolink amplification kit (Leica, Newcastle Inc., UK) was used for signal amplification, and 3.3′-diaminobenzidine tetrachloride (0.25 mg dissolved in 1 mL 0.02% hydrogen peroxide) was used as a precipitating substrate for signal detection. The specificity of the primary antibodies was confirmed by the appropriate reagent controls (omitting the primary antibody or substituting non-immune serum for the primary antibody in the staining protocol), which revealed no staining. The positive expression of AGTR-1, AGTR-2 and D2-40 was indicated by brown cytoplasmic staining of the cells.
HistomorphometryThe area fraction occupied by AGTR-1/AGTR-2+ cells in the alveolar septae and vascular walls and lymphatic vessels along the peripheral and periaxial interstitium was determined with a point-counting procedure (38). We used a 100-point grid with a known area (187.500 μm2 at 400× magnification) attached to the microscope eyepiece (38). Ten fields were chosen from across multiple sites, accounting/adjusting for the cellularity or number of positively immunostained cells per connective tissue area (38). Thus, the septal area in each field was determined according to the number of points falling within the positive cells in the field of view (as a proportion of the total grid area). Afterwards, the number of positive cells within the septal area was counted. The immunostaining cellularity was determined as the number of positive cells in each field divided by the septal area. The final results were expressed as the mean±standard deviation (SD) of the lung specimens for each patient in 10 random, non-coincident microscopic fields.
To control for variation in the scoring between our 2 histologists (ERP and VLC), 20% of the stained slides were independently scored by both observers. The coefficient of variance between the cell counts for the 2 observers was <5%.
Statistical analysisThe data are reported as the means±SD. The statistical analyses were performed using an analysis of variance (ANOVA) followed by the appropriate post hoc tests, including Bonferroni correction and Student's t-test for comparisons between 2 groups. The survival curves were determined using the Kaplan-Meier method, and the risk of death was estimated using a Cox regression analysis. The statistical program SPSS, Inc. (USA) was used, and p<0.05 was considered to be significant.
RESULTSClinical featuresThe clinical features of the 56 patients included in this study are shown in Table1. All studied patients showed a restrictive lung function pattern that was characterized by a decrease in TLC (the mean values were 81% of that predicted for SSc-NSIP and 78% of that predicted for IPF-UIP) and an increased FEV 1/FVC ratio/100 (the mean value was 107% of that predicted for SSc-NSIP vs. 90% of that predicted for IPF-UIP). The mean predicted DLCO values were significantly decreased in the IPF-UIP patients (55%) compared with the SSc-NSIP (66%) patients, and they were significantly greater when comparing the DLCO/VA in the IPF-UIP (47%) vs. the SSc-NSIP (77%) patients. No difference was found in the FEV1, FCV, TLC and RV values between the SSc-NSIP and IPF-UIP groups (Table1).
Clinical data of the patients with systemic sclerosis and idiopathic pulmonary fibrosis.
| SSc-NSIP (n = 30) | IPF-UIP (n = 26) | p-value | |
|---|---|---|---|
| Age, years | 45±8 | 64±7 | <0.001 |
| Sex (female/male) | 30 | 10/16 | 1.2 |
| Spirometry | |||
| FEV1 (% predicted) | 70±13.8 | 76±20.01 | 0.23 |
| FVC (% predicted) | 65±13.8 | 69±16.8 | 0.35 |
| FEV1/FVC | 107±8.69 | 90±18.7 | <0.001 |
| TLC (% predicted) | 81±11.5 | 78±21.9 | 0.64 |
| RV (% predicted) | 117±35.5 | 101±62.04 | 0.36 |
| DLCO (% predicted) | 66±21.6 | 55±19.1 | 0.01 |
| DLCO/VA (% predicted) | 77±35.2 | 47±22.74 | 0.008 |
The data are expressed as the means±SD or as counts (percentages); p<0.05 was significant.
Abbreviations: NSIP, non-specific interstitial pneumonia; UIP, usual interstitial pneumonia; FEV1, forced expiratory volume in 1 s; FVC, forced vital capacity; TLC, total lung capacity; RV, residual volume; DLCO, diffusing capacity of the lung for carbon monoxide; VA, alveolar volume.
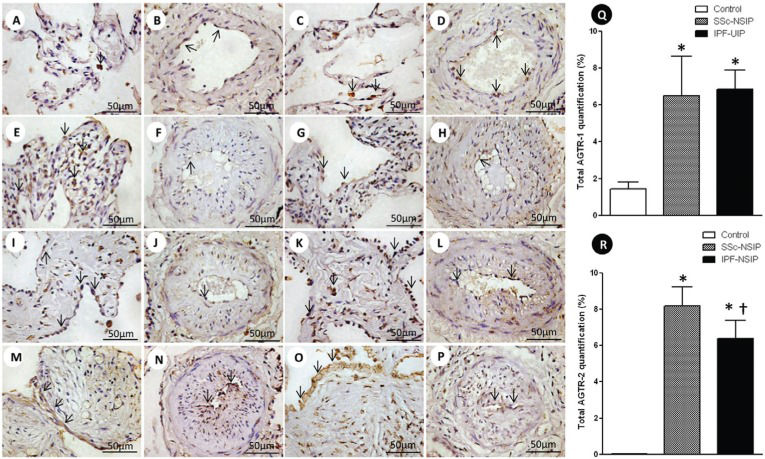
Alveolar septae and vessels from the normal control lungs, NSIP and UIP histologic patterns are shown in Figure1 (immunostained with the AGTR-1 and AGTR-2 antibodies). Different immunostaining intensity was observed in the epithelial, endothelial, myofibroblast and smooth muscle cells from the alveolar septae and vessels in the SSc-NSIP (cellular SSc-NSIP and fibrotic SSc-NSIP) and IPF-UIP groups compared to the normal control lungs. Table2 summarizes the morphometric results. A significantly higher percentage of the septal and vessel cells immunostained for AGTR-1/AGTR-2 in the cellular SSc-NSIP, fibrotic SSc-NSIP and IPF-UIP lungs was observed compared to the controls. In the SSc-NSIP and IPF-UIP lungs, a similar proportion of the septal cells immunostained for AGTR-1 was observed, except for the IPF-UIP lungs, in which the AGTR-2 immunoreactivity represented a significantly reduced proportion of the septal cells compared to the SSc-NSIP lungs. Of equal significance was the increased percentage of the vessel cells that were immunostained for AGTR-1 in the cellular SSc-NSIP and IPF-UIP lungs. Concerning AGTR-2, a similar percentage of the vessel cells was present in the fibrotic SSc-NSIP and IPF-UIP lungs. When we compared the total cellular expression of AGTR-1 in the SSc-NSIP and IPF-UIP groups, a similar proportion was observed; however, both were increased compared to the control group (Figure Q), except for the IPF-UIP lungs, in which the total cellular AGTR-2 expression had a significantly smaller proportion than that of the SSc-NSIP pattern lungs (Figure R).
 and AGTR-2 divided in septal areas and intrapulmonary vessels from normal control lungs, systemic sclerosis (SSc) and idiopathic pulmonary fibrosis (IPF). In (A) and (B), we observe a diffuse and minimal AGTR-1 (arrows) expression in the alveolar septal areas and in the intrapulmonary vessels of the controls. The AGTR-1 expression shows greater increases in the thickened alveolar septal region from the cellular SSc-NSIP (arrows, E) than in the fibrotic SSc-NSIP (arrows, I). In the intrapulmonary vessels, the expression of AGTR-1 (arrows) is minimal and similar in both groups (cellular SSc-NSIP, F and fibrotic SSc-NSIP, J). Increased expression of AGTR-1 (arrows) is observed in the septal areas (M) and intrapulmonary vessels (N) of IPF-UIP. In (C) and (D), we observed AGRT-2 (arrows) expression in the septal areas and intrapulmonary vessels from the controls. In (G) and (K), we observed a similar expression of AGTR-2 (arrows) in the septal areas of cellular and fibrotic SSc-NSIP. Increased expression of AGTR-2 (arrows) is observed in the intrapulmonary vessels of fibrotic SSc-NSIP (L) compared to cellular SSc-NSIP (H). The expression of AGTR-2 (arrows) is more evident in the septal areas of IPF-UIP (O) than in the intrapulmonary vessels (P) of this histologic pattern. Q and R show the total values of AGTR-1 and AGTR-2 and the results analyzed by a one-way ANOVA with the Bonferroni multiple comparisons test for the control, SSc-NSIP and IPF-UIP groups. (*) Significantly higher AGTR-1 expression was observed in the SSc-NSIP and IPF-UIP lungs compared with the control group (p =0.001 and p =0.001, respectively). A similar situation was observed with AGTR-2 expression from the SSc-NSIP and IPF-UIP patterns compared to the control group (p<0.001 and p<0.001, respectively). (†) Significantly higher expression of AGTR-2 in the SSc-NSIP group was observed when compared with the IPF-UIP group (p = 0.04) by Student")
Cellular expression of the angiotensin II type 1 receptor (AGTR-1) and AGTR-2 divided in septal areas and intrapulmonary vessels from normal control lungs, systemic sclerosis (SSc) and idiopathic pulmonary fibrosis (IPF). In (A) and (B), we observe a diffuse and minimal AGTR-1 (arrows) expression in the alveolar septal areas and in the intrapulmonary vessels of the controls. The AGTR-1 expression shows greater increases in the thickened alveolar septal region from the cellular SSc-NSIP (arrows, E) than in the fibrotic SSc-NSIP (arrows, I). In the intrapulmonary vessels, the expression of AGTR-1 (arrows) is minimal and similar in both groups (cellular SSc-NSIP, F and fibrotic SSc-NSIP, J). Increased expression of AGTR-1 (arrows) is observed in the septal areas (M) and intrapulmonary vessels (N) of IPF-UIP. In (C) and (D), we observed AGRT-2 (arrows) expression in the septal areas and intrapulmonary vessels from the controls. In (G) and (K), we observed a similar expression of AGTR-2 (arrows) in the septal areas of cellular and fibrotic SSc-NSIP. Increased expression of AGTR-2 (arrows) is observed in the intrapulmonary vessels of fibrotic SSc-NSIP (L) compared to cellular SSc-NSIP (H). The expression of AGTR-2 (arrows) is more evident in the septal areas of IPF-UIP (O) than in the intrapulmonary vessels (P) of this histologic pattern. Q and R show the total values of AGTR-1 and AGTR-2 and the results analyzed by a one-way ANOVA with the Bonferroni multiple comparisons test for the control, SSc-NSIP and IPF-UIP groups. (*) Significantly higher AGTR-1 expression was observed in the SSc-NSIP and IPF-UIP lungs compared with the control group (p =0.001 and p =0.001, respectively). A similar situation was observed with AGTR-2 expression from the SSc-NSIP and IPF-UIP patterns compared to the control group (p<0.001 and p<0.001, respectively). (†) Significantly higher expression of AGTR-2 in the SSc-NSIP group was observed when compared with the IPF-UIP group (p = 0.04) by Student's t-test.
Summary of the morphometric resultsa.)
| Variables | Control | Cellular SSc-NSIP | Fibrotic SSc-NSIP | SSc-NSIP | UIP |
|---|---|---|---|---|---|
| Septal AGTR-1 | 1.45±1.02 | 10.72±6.20 | 12.18±8.00 | 11.41±6.97 | 10.10±22.23 |
| Vascular AGTR-1 | 1.06±1.03 | 4.36±5.35 | 0.51±1.15 | 2.53±4.33 | 2.13±1.64 |
| Septal AGTR-2 | 0.03±0.02 | 12.75±8.05 | 12.78±6.96 | 12.77±7.36 | 7.78±4.31 |
| Vascular AGTR-2 | 0.01±0.04 | 1.26±1.35 | 6.91±4.64 | 3.83±4.30 | 6.06±4.66 |
| Total AGTR-1 | 1.25±0.84 | 7.54±4.49 | 6.34±3.96 | 6.97±4.18 | 6.26±10.93 |
| Total AGTR-2 | 0.02±0.02 | 6.51±3.88 | 9.85±5.09 | 8.18±4.73 | 6.38±3.63 |
| Lymphatic density | 1.68±0.38 | 2.72±0.62 | 2.92±0.69 | 2.80±0.65 | 3.73±1.19 |
| Lymphatic area | 2.81±0.90 | 4.28±1.32 | 4.33±0.64 | 4.25±1.08 | 5.52±2.24 |
a The units of “% of points” indicate the number of points overlying the phenomena of interest divided by the total number of points overlying the septae and vessels. In morphometry, this process is called a point fraction and is often symbolized as Pp. The Pp has been shown to approximate the volume fraction or Vv.
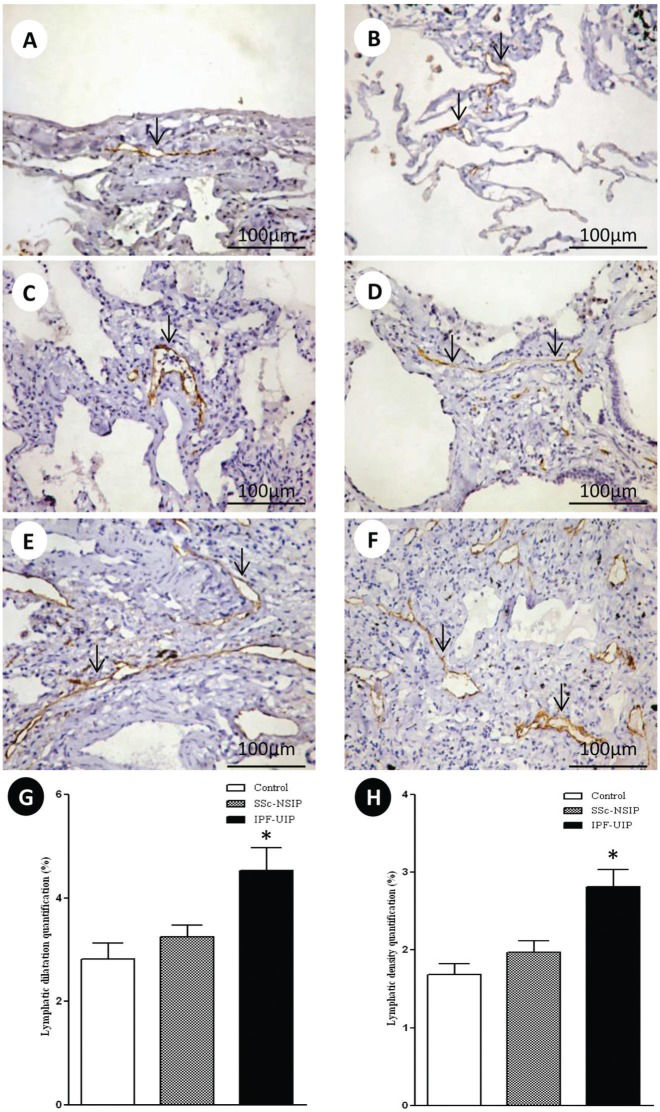
Figure2 shows the peripheral, interlobular and peribronchovascular lymphatic vessels from the normal control lungs and the SSc-NSIP and IPF-UIP histologic patterns, immunostained for D2-40. A different proportion of the lymphatic vessels in the peripheral, interlobular and peribronchovascular interstitium was visualized in the SSc-NSIP (cellular SSc-NSIP and fibrotic SSc-NSIP) and IPF-UIP lungs compared to the control lungs. Table2 summarizes the morphometric results. In the peripheral, interlobular and peribronchovascular interstitium of the IPF-UIP group, the lymphatic vessels showed a significantly increased area compared to the SSc-NSIP lungs (Figure G). A significantly higher percentage of lymphatic vessels was present in the peripheral, interlobular and periaxial interstitium of the IPF-UIP pattern compared to the SSc-NSIP histologic pattern (Figure H).
 and idiopathic pulmonary fibrosis (IPF). The lymphatic vessels (arrows) were observed in the sub pleural (A) and interlobular septal interstitium (B) in the control lungs. In cellular SSc-NSIP, the lymphatic vessels (arrows) were observed in the thickened alveolar septal region (C) and were infrequent compared to the fibrotic SSc-NSIP pattern (arrows, D). In the UIP pattern, we observed a higher proportion of lymphatic vessels (arrows) in the interlobular and subpleural spaces (E and F). G and H represent the total values of the lymphatic dilatation and density, and the results analyzed by the one-way ANOVA followed by the Bonferroni multiple comparisons test for the control, SSc-NSIP and IPF-UIP groups. (*) A significantly increased lymphatic dilatation (G) was observed in the IPF-UIP group compared to the SSc-NSIP (p = 0.01) and control (p = 0.01) groups. Similarly, a significantly higher lymphatic density (H) was observed in the IPF-UIP group compared to the SSc-NSIP (0.02) and control (0.01) groups.")
D2-40 cell expression in the lymphatic vessels of the normal control lungs, systemic sclerosis (SSc) and idiopathic pulmonary fibrosis (IPF). The lymphatic vessels (arrows) were observed in the sub pleural (A) and interlobular septal interstitium (B) in the control lungs. In cellular SSc-NSIP, the lymphatic vessels (arrows) were observed in the thickened alveolar septal region (C) and were infrequent compared to the fibrotic SSc-NSIP pattern (arrows, D). In the UIP pattern, we observed a higher proportion of lymphatic vessels (arrows) in the interlobular and subpleural spaces (E and F). G and H represent the total values of the lymphatic dilatation and density, and the results analyzed by the one-way ANOVA followed by the Bonferroni multiple comparisons test for the control, SSc-NSIP and IPF-UIP groups. (*) A significantly increased lymphatic dilatation (G) was observed in the IPF-UIP group compared to the SSc-NSIP (p = 0.01) and control (p = 0.01) groups. Similarly, a significantly higher lymphatic density (H) was observed in the IPF-UIP group compared to the SSc-NSIP (0.02) and control (0.01) groups.
A significant inverse association was found between the vascular AGTR-2 and DCLO/VA (R = -0.74, p =0.001) in the SSc-NSIP and (R = -0.78, p =0.001) UIP groups. Equally significant was the negative association between the lymphatic vessel area in SSc-NSIP and the predicted values of DLCO (R = -0.53, p =0.04).
Survival analysisNineteen patients died during the 180-month follow-up period (5 SSc-NSIP and 14 IPF-UIP). A preliminary examination of the Kaplan-Meier survival curves showed that in this study, the patients with the SSc-NSIP pattern with percentages of vessel cells immunostained for AGTR-1 that were <2.49% and AGTR-2 percentages <7.46% had higher survival rates than the patients with percentages of vessel cells immunostained for AGTR-1 that were >2.49% and AGTR-2 percentages >7.46. The multivariate analyses by the Cox regression model showed statistical significance (log-likelihood = 59.39, p<0.0001) with a high risk of death for the patients with the IPF-UIP pattern (p =0.01) and a high percentage of vessel cells immunostained for the AGTR-2 and lymphatic vessels (Table3).
Cox proportional hazards regression to ascertain the individual contribution of the histological patterns (NSIP and UIP) and the morphological factors associated with survival and to compare the adjusted survival between the groups (-2 log-likelihood = 59.93; Chi-squared = 40.10; p<0.001).
| β | SE | Wald test | p-value | Exp (β) | 95.0% CI for Exp (β) | ||
|---|---|---|---|---|---|---|---|
| Lower | Upper | ||||||
| NSIP pattern | -12.70 | 182.18 | 5.97 | 0.94 | 0.00 | 0.00 | 0.60E149 |
| UIP pattern | -1.60 | 0.65 | 5.97 | 0.01 | 0.20 | 0.56 | 0.78 |
| AGTR2 vascular | 0.26 | 0.89 | 9.23 | 0.002 | 1.30 | 1.10 | 1.55 |
| Lymphatic density | -1.20 | 0.25 | 6.92 | 0.008 | 0.297 | 0.122 | 0.73 |
β = beta coefficient; SE = standard error; Exp (β) = exponential beta; CI = Confidence interval.
Unlike SSc-NSIP, the likely reason that IPF-UIP patients do not respond to immunosuppressive therapies and their clinical course is marked by inexorable deterioration is the imbalance between parenchymal and vascular remodeling. Therefore, it is important to determine whether additional histological details can help us understand the clinical differences between these diseases. After pulmonary injury, parenchymal and vascular remodeling contribute to increased pulmonary epithelial and endothelial permeability. This early phase is followed by a subacute fibroproliferative phase, which may allow repair of the injured lung or may result in progressive obliteration of the interstitial, alveolar and vascular compartments of the lung through a fibroproliferative process that may be established by 24 h after the injury (39).
Recent studies have shown that ANG II contributes to pulmonary fibrosis progression, which is evidenced not only by its potent vasoconstrictor activity but also by its influence on organ remodeling and fibrosis (20,21,22). The AGTR-1 and AGTR-2 binding of ANG II is facilitated by the cleavage of ANG I by the ACE. Both receptors belong to the 7-transmembrane G protein–coupled receptor family (23,24). Whereas most of the ANG II-mediated effects, such as cell growth, inflammation or extracellular matrix synthesis, have been shown to be mediated via AGTR-1, the function of AGTR-2 has been less clearly investigated (40,41). During the development of fibrosis, AGTR expression undergoes an expression shift in favor of higher AGTR-2 levels, thereby tilting the cellular response to ANG II (42-44). Thus, for all these reasons, we should not be surprised to learn that immunohistochemical staining for AGTRs provides important information about the organ remodeling process in diffuse parenchymal lung diseases, and our results now confirm the pathogenetic importance of AGTRs in IPF and SSc.
We found a significantly greater percentage of septal and vessel cells immunostained for AGTR-1 in SSc-NSIP and IPF-UIP compared with the normal lung. In addition, SSc-NSIP and IPF-UIP had a similar proportion of septal cells immunostained for AGTR-1. We also found an increased percentage of vessel cells immunostained for AGTR-1 in cellular SSc-NSIP and IPF-UIP. Previous reports in the literature have shown the potential role of ANG II in experimental lung fibrosis and in IPF (42-44), while the potency of AGTR-1 in fostering lung fibrosis has been previously demonstrated in multiple in vitro cell culture conditions as well as in vivo animal models (45,42). In experimental animal models of lung fibrosis, using bleomycin exposure or radiation, the fibrotic response was attenuated by the ACE inhibitors and by specific AGTR-1 antagonism (45,46). Studies of the inhibition of the ANG II pathway have demonstrated opposing results. Some studies have shown beneficial results and blocked the fibrosis, principally in bleomycin-induced pulmonary fibrosis (47); others have failed to document a beneficial effect of the ANG II blockade in experimental models of lung fibrosis, indicating that the fibrotic disease process is dependent not only on AGTR-1 activity (48). We found that a small percentage of vessel cells in SSc-NSIP are AGTR-1+. In contrast to the data available for AGTR-1, the function of AGTR-2 is much less well explored. In our study, there was an increased percentage of vessel cells immunostained for AGTR-2 in SSc-NSIP and IPF-UIP, suggesting an important role in the pathogenesis of these disorders. In heart tissue, the AGTR-2 is considered to be a neutralizer, modulating the actions of AGTR-1 (28,29), but many signaling mechanisms and receptor functions are unclear, particularly the role of AGTR-2 in pulmonary fibrosis.
We also found a significantly higher percentage of lymphatic vessels in the peripheral, interlobular and periaxial interstitium of IPF-UIP compared with the SSc-NSIP histologic pattern. In IPF-UIP, the lymphatic vessels showed a significantly increased area compared with SSc-NSIP. The lymphatic vessels contribute to fibrosis maturation and scar formation through the drainage of excessive proteins and fluid during fibrosis (49). In the present study, D2-40+ lymphatics were abundant in the peripheral, interlobular and peribronchovascular interstitium. Previous studies performed by our group have shown that the lymphatic vessels play an important role in early remodeling in the development of pulmonary fibrosis (50). The current study clearly demonstrated that the lymphatic vessels were poorly detected in the affected areas of early remodeling, such as cellular SSc-NSIP, compared to the fibrotic SSc-NSIP and IPF-UIP patterns.
Our study has clinical and functional importance. A significant inverse association was found between vascular AGTR-2 and DCLO/VA in the SSc-NSIP and IPF-UIP groups. Of equal significance was the negative association between the lymphatic vessel area in SSc-NSIP and the predicted values of DLCO. To establish the relevance of these findings to disease progression in the patients, the AGTRs were evaluated as a function of survival (controlled for age) in the IPF-UIP and SSc-NSIP histologic patterns. The multivariate analyses using the Cox regression model showed a high risk of death for the patients with the IPF-UIP pattern presenting a high percentage of vessel cells immunostained for AGTR-2 in the lymphatic vessels.
We concluded that the AGTRs and lymphatic vessels in the lung parenchyma offer the potential to control the organ remodeling and fibrosis involved in SSc-NSIP and IPF-UIP progression, which suggests that strategies aimed at preventing high AGTR synthesis and low lymphangiogenesis may have a greater impact on SSc and IPF. Further study using a randomized and prospective trial is necessary to finalize this conclusion.
AcknowledgmentsWe are grateful to Sandra de Morais Fernezlian, a biologist from the immunohistochemistry laboratory, for the helpful immunohistochemistry staining results and to Prof. Carlos Roberto Ribeiro de Carvalho and Ronaldo Adib Cairalla, from the Department of Pneumology, for their patient databases. This study was supported by the National Council for Scientific and Technological Development (CNPq) and the Foundation for the Support of Research of the State of São Paulo (2008/53022-3 and 2011/06312-9).
AUTHOR CONTRIBUTIONSAll authors designed the study, analyzed and interpreted the data and wrote the manuscript.
No potential conflict of interest was reported.