



The objective of this study was to determine the effect of nonspecific phosphodiesterase inhibition on transcription factor activation and tumor necrosis factor-alpha (TNF-α) production in lipopolysaccharide (LPS)-stimulated human mononuclear cells.
INTRODUCTIONThe production of TNF-α following LPS stimulation is one of the key steps in bacterial sepsis and inflammation. The mechanism by which phosphodiesterase inhibition alters TNF-α production in the presence of LPS remains unclear.
METHODSHuman mononuclear cells were stimulated with LPS (1 μg/mL), in the presence and absence of Pentoxifylline (PTX; 20 mM), a nonspecific phosphodiesterase inhibitor. Western blotting of phosphorylated cytoplasmic I-κBα, nuclear factor-κB p65 (NF-κB), and nuclear cAMP-response element binding protein (CREB) was performed. DNA binding of NF-κB and CREB was verified by electrophoretic mobility shift assay. TNF-α levels were determined in the supernatant of stimulated cells in the presence and absence Protein kinase A inhibition by an enzyme-linked immunosorbent assay (ELISA).
RESULTSPTX was demonstrated to significantly reduce cytoplasmic I-κBα phosphorylation, nuclear p65 phosphorylation, and the DNA binding activity of NF-κB. In contrast, PTX markedly enhanced the phosphorylation and DNA binding activity of CREB. Cells concomitantly treated with PTX and LPS secreted similar levels of TNF-α in the presence and absence Protein kinase A inhibition.
DISCUSSIONThe increased level of cAMP that results from phosphodiesterase inhibition affects cytoplasmic and nuclear events, resulting in the attenuation of NF-κB and the activation of CREB transcriptional DNA binding through pathways that are partially Protein kinase A-independent.
CONCLUSIONPTX-mediated phosphodiesterase inhibition occurs partially through a Protein kinase A-independent pathway and may serve as a useful tool in the attenuation of LPS-induced inflammation.
The bacterial membrane component lipopolysaccharide (LPS) is capable of initiating phosphorylation and activation of multiple host intracellular protein kinases and transcription factors. Transcription factor activation, which results in the modulation of gene transcription and protein synthesis, is a critical element in the defense mechanism of the host immune system.1,2
LPS-induced transcription factor activation has been shown to be a key regulator of tumor necrosis factor-α (TNF-α) production.3 TNF-α is a potent pro-inflammatory cytokine involved in a wide spectrum of cellular responses. Furthermore, TNF-α synthesis can be attenuated in immune cells exposed to phosphodiesterase (PDE) inhibition after challenge by a variety of pro-inflammatory stimulants.4 The signaling mechanisms affected by PDE inhibition, which ultimately lead to the downregulation of TNF-α production, have not been well characterized in inflammatory cells.
Classically, it has been demonstrated that PDE inhibition results in the intracellular accumulation of the second messenger cyclic adenosine-3,5-monophosphate (cAMP) and subsequent activation of Protein kinase A (PKA).5 PKA activation then leads to the phosphorylation of the transcription factor cAMP-response element binding protein (CREB), transmission of signals into the nucleus, and the subsequent modulation of gene transcription.6 This apparently simple linear cascade does not fully explain the mechanism by which an elevation in the intracellular cAMP level exerts wide-ranging effects on multiple cellular functions. There is a growing body of evidence suggesting that cAMP may function through both PKA-dependent and -independent mechanisms.6–8
LPS-induced activation of the transcription factor nuclear factor-κB (NF-κB) has also been the focus of a great deal of research. It has been clearly demonstrated that agents that increase intracellular cAMP also inhibit NF-κB-dependent pro-inflammatory gene transcription, particularly of the TNF-α gene.9
Controversy exists regarding the exact mechanism(s) by which PDE inhibition down-regulates TNF-α production. Possibilities include, but are not limited to, inhibition of NF-κB DNA binding activity, downregulation of NF-κB transcriptional activity, increased CREB activation, and competition between NF-κB and CREB for common co-activators such as CREB binding protein (CBP). It is also not known whether these processes rely solely upon the activation of PKA.
Therefore, the objective of the present study is to determine the effects of nonspecific PDE inhibition with 1-[5-oxohexyl]-3,7-dimethylxanthine (Pentoxifylline; PTX) on NF-κB and CREB activation in vitro in human mononuclear cells. With the use of specific inhibitors, we also investigated the role of PKA in LPS-induced TNF-α production.
MATERIALS AND METHODSThis study was approved by the Human Research Protections Program and the Institutional Review Board. Written consent to participate in the study was obtained from all volunteers prior to blood donation.
Heparinized Vacutainers were purchased from Becton Dickinson (San Jose, CA). 1.5-mL Eppendorf centrifuge tubes were purchased from Fisher Scientific (Pittsburg, PA). Hank’s balanced salt solution (HBSS) and RPMI 1640 were obtained from Irvine Scientific (Santa Ana, CA). LPS from Escherichia coli serotype 0111:B4 and PTX were purchased from Sigma (St. Louis, MO). The PKA inhibitor, N-[2-((p-bromocinnamyl)amino)ethyl]-5-isoguinolinesulfonamide-2HCl] (H89), was purchased from Calbiochem (La Jolla, CA). Antibodies for phosphorylated CREB (serine 133), phosphorylated I-κBα (serines 32 and 36), phosphorylated NF-κB p65 subunit (serine 276), and secondary antibodies were purchased from Cell Signaling (Beverly, MA).
Dextran T500 and Percoll were received from GE Healthcare/Amersham Biosciences (Piscataway, NJ). The enzyme-linked immunosorbent assay (ELISA) for TNF-α was obtained from Quantikine R&D Systems (Minneapolis, MN).
Tris-glycine and DNA retardation gels, nitrocellulose membranes, and western blot running buffers were obtained from Invitrogen (Carlsbad, CA). NE-PER® nuclear and cytoplasmic extraction reagents, the BCA protein reagent kit, the Supersignal West Pico Chemiluminescent Kit, and the LightShift Chemiluminescent EMSA Kit were purchased from Pierce (Rockland, IL). CREB and NF-κB DNA oligonucleotide probes were synthesized by IDT (Coralville, IA). Nylon membranes were purchased from Roche Applied Sciences (Indianapolis, IN). Western blot band quantification was performed with the UN-SCAN-IT Gel Digitizing software (Silk Scientific, Orem, UT).
Human Peripheral Blood Mononuclear Cell Isolation and StimulationHuman mononuclear cells were isolated from the peripheral blood of four healthy human volunteers. The sample size was chosen from our previously published work, which demonstrated reliable results.10 Cell isolation and all subsequent experiments were conducted under sterile and pyrogen-free conditions. Blood collected in heparin tubes was incubated with Dextran T500 and the red cells were sedimented for 40 minutes at room temperature. The resultant serum was then washed with HBSS and separated by Percoll gradient centrifugation according to the manufacturer’s instructions. Cell viability was assessed by trypan blue dye exclusion, with purity of greater than 95%. Isolated cells were resuspended in RPMI 1640 supplemented with 10% FBS and 5 mM HEPES at a concentration of 1 x 107cells/mL. Each subsequent experiment listed below was conducted on samples of 5 x 106 cells per treatment group. Isolated cells were stimulated with either HBSS as a negative control, LPS (1 μg/mL), PTX (20 mM), or concomitant LPS / PTX, at the above-mentioned concentrations, for 30 minutes at 37ºC. Cells were placed on ice for 10 minutes to stop the reaction and the samples were then stored at −70°C for further analysis. The stimulation times and concentrations of LPS and PTX utilized in this study were determined in previous pilot studies done in our laboratory, which examined the effects of increasing concentrations of PTX on LPS-induced TNF-α production. LPS at a concentration of 1 μg/mL and PTX at a concentration of 20 mM were the minimum concentrations necessary to produce a reproducible and reliable up-regulation and down-regulation in TNF-α production, respectively, in quantitative assays (data not shown).
Effect of PTX on LPS-induced TNF-α production by mononuclear cellsThe concentration of TNF-α was quantitatively determined by ELISA in the media of cells exposed to the treatments described above after stimulation for up to a maximum of 18 hours, at 3-hour intervals. The results are expressed in pg/mL.
SDS-PAGE and Immunoblotting (I-κBα, NF-κB p65, and CREB)Following stimulation, PBMCs were washed with ice-cold PBS and centrifuged to collect a cell pellet. The pellet was resuspended in ice-cold SDS sample buffer supplemented with 100 mM dithiothreitol. Cytoplasmic and nuclear extracts were isolated with NE-PER nuclear and cytoplasmic extraction reagents with 1x Halt Protease Inhibitor Cocktail according to the manufacturer’s instructions. Protein concentrations were determined with the BCA protein reagent kit for each sample according to a standardized curve for albumin. Cell lysates were collected by boiling the samples in a 100ºC water bath for 5 minutes. Ten μg of protein per sample was separated by SDS-polyacrylamide gel electrophoresis through 8–16% tris-glycine polyacrylamide gradient gels and transferred to nitrocellulose membranes. The membranes were blocked with 5% milk in Tris-buffered saline/Tween 20 (Fischer Scientific, Pittsburgh, PA) for 1 hour. Cytoplasmic extracts were incubated with phosphorylated I-κBα antibody (1:200), while nuclear extracts were incubated with either phosphorylated NF-κB p65 antibody (1:500) or phosphorylated CREB antibody (1:500) overnight at 4°C in separate experiments. The membranes were washed with Tris-buffered saline/Tween 20 and incubated for 1 hour at room temperature with the secondary antibody, horseradish peroxidase-linked anti-rabbit IgG diluted 1:2000 in blocking solution. After repeated washing of the membrane, the Supersignal West Pico Chemiluminescent Kit was applied for antibody detection per the manufacturer’s instructions.
Western blot data is presented as a percentage of LPS stimulation. The percentage of LPS stimulation was calculated by dividing the mean band pixel total for each treatment arm divided by the mean pixel total of samples stimulated with LPS and multiplying by 100. Thus, LPS stimulation is reported as 100% and each of the treatment arms is reported as a percent of LPS stimulation.
NF-κB and CREB Electrophoretic Mobility Shift Assay (EMSA)The non-radioactive LightShift Chemiluminescent EMSA Kit was used to detect DNA-protein interactions. The NF-κB 3′ biotin end-labeled DNA oligonucleotide used as a probe for the EMSA was a 42-bp double stranded construct (5′-TTGTTACAA-GGGGACTTTCCGCTGGGGACTTTCCAGGGAGGC – 3′) containing two tandemly repeated NF-κB binding sites (underlined). Specificity was determined by a competition assay with the addition of 200 molar excess of unlabeled double stranded NF-κB oligonucleotide. The CREB 3′ biotin end-labeled DNA oligonucleotide used as a probe for the EMSA was a 23-bp double stranded construct (5′-TTT TCG AGC TCTGACGTCAGA-GC – 3′) containing the CRE consensus sequence (underlined). Specificity was determined by a competition assay with the addition of 200 molar excess of unlabeled double stranded CREB oligonucleotide.
Nuclear extracts (10 μg) were incubated with 5 nM NF-κB or CREB probe (NF-kB: 1x binding buffer, 50 mM KCl, 1 mM EDTA, 1 mM DTT, 0.1% NP40, 10% glycerol, and 50 ng/μl poly dI-dC, CREB: 1x binding buffer, 20 mM Tris, pH 7.5, 50 mM KCl, 1 mM EDTA, 1 mM DTT, 0.10% NP40, 6% glycerol, 0.1 mg/mL BSA and 50 ng/μl poly dI-dC) and were then electrophoresed through a 6% DNA retardation gel at 100V for 90 minutes. The gels were electrophoretically transferred at 380mA for 1 hour on ice to a positively charged nylon membrane and immediately cross-linked for 15 minutes with a UV transilluminator equipped with a 312 nm bulb. Streptavidin-horseradish peroxidase conjugate and the LightShift Chemiluminescent Substrate were used to detect the biotin end-labeled DNA. The nylon membranes were exposed to x-ray film for 1–3 minutes for detection.
Role of PKA on LPS-induced mononuclear cell TNF-α productionTo evaluate the role of PKA on TNF-α production in mononuclear cells and its involvement in the attenuation of LPS-induced TNF-α production observed following PTX treatment, H89, a specific PKA inhibitor, was utilized. Isolated mononuclear cells were incubated according to the treatment groups described above in the presence and absence of pretreatment with H89 (10 μM) for 1 hour at 37ºC. The dose of H89 was chosen based on previous work which demonstrated specific and complete inhibition of PKA at this concentration.10 Since the activity of this inhibitor is both specific and consistent at this specified dose, we did not include experiments with additional PKA inhibitors with this set of experiments. The TNF-α concentration in the supernatant was measured quantitatively by ELISA. The results are expressed in pg/mL.
Statistical AnalysisThe experimental results obtained in this study were derived from four separate experiments with healthy volunteer donors. Each assay was performed in duplicate or triplicate where appropriate. Data is presented as the mean ± SEM. Statistical differences between groups were determined by one-way analysis of variance (ANOVA) with a Bonferroni correction. Statistical significance was defined as P < 0.05.
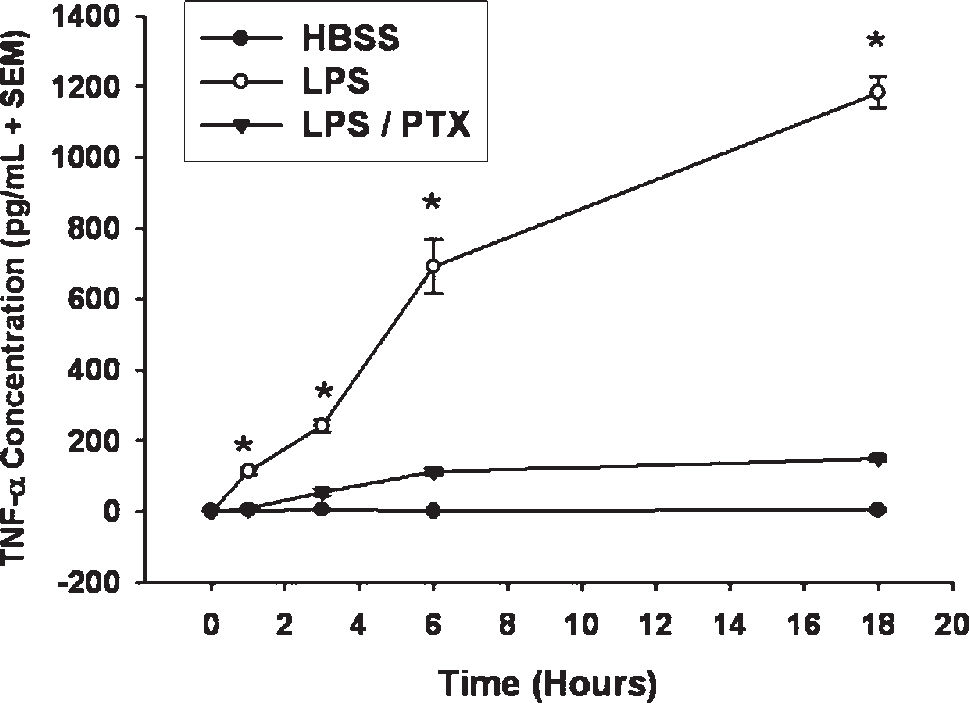
RESULTSThe effects of PTX on LPS-induced TNF-α productionTo determine the duration of PDE inhibition on TNF-α production after LPS stimulation, time course studies were conducted. In accordance with prior work, PTX had a rapid and sustained effect on TNF-α production in vitro (Figure 1).
 were incubated with LPS (1 μg/mL) in the presence and absence of Pentoxifylline (PTX; 20 mM). TNF-α concentrations in supernatant were measured by ELISA (pg/mL) up to 18 hours after stimulation (n = 4 per group). PTX downregulated TNF-α production by mononuclear cells in a rapid and sustained manner (*, P < 0.05 vs. LPS / PTX).")
Phosphodiesterase inhibition attenuates LPS-induced TNF-α production. Isolated human mononuclear cells (5 x 106) were incubated with LPS (1 μg/mL) in the presence and absence of Pentoxifylline (PTX; 20 mM). TNF-α concentrations in supernatant were measured by ELISA (pg/mL) up to 18 hours after stimulation (n = 4 per group). PTX downregulated TNF-α production by mononuclear cells in a rapid and sustained manner (*, P < 0.05 vs. LPS / PTX).
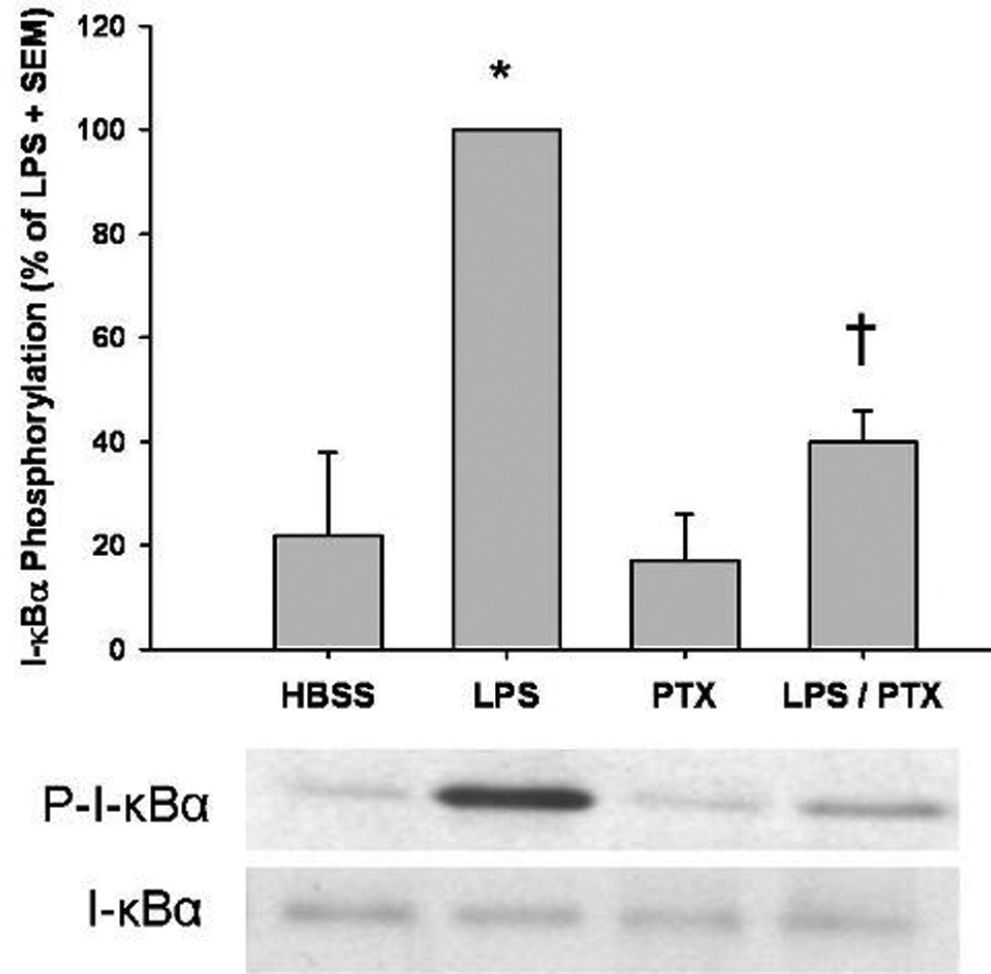
Since phosphorylation leads to ubiquitination and degradation of I-κBα and subsequent nuclear translocation of NF-κB, we first examined the effects of PTX on I-κBα. Cytoplasmic I-κBα phosphorylation was markedly increased following LPS stimulation when compared to control (100 ± 0 vs. 20 ± 18; P < 0.05). The addition of PTX significantly downregulated LPS-induced I-κBα phosphorylation (P = 0.02; Figure 2)
 were incubated with LPS (1 μg/mL) and Pentoxifylline (PTX; 20 mM). I-κBα phosphorylation was assessed by western blotting, as described in Materials and Methods. Results are expressed as a percentage of LPS stimulation ± SEM (n = 4 per group). LPS-stimulation markedly enhanced I-κBα phosphorylation over that of the control (*, P < 0.05 vs. HBSS). The addition of PTX resulted in a marked reduction in I-κBα phosphorylation (†, P = 0.02 vs. LPS).")
Phosphodiesterase inhibition attenuates LPS-induced I-κBα phosphorylation. Isolated human mononuclear cells (5 x 106) were incubated with LPS (1 μg/mL) and Pentoxifylline (PTX; 20 mM). I-κBα phosphorylation was assessed by western blotting, as described in Materials and Methods. Results are expressed as a percentage of LPS stimulation ± SEM (n = 4 per group). LPS-stimulation markedly enhanced I-κBα phosphorylation over that of the control (*, P < 0.05 vs. HBSS). The addition of PTX resulted in a marked reduction in I-κBα phosphorylation (†, P = 0.02 vs. LPS).
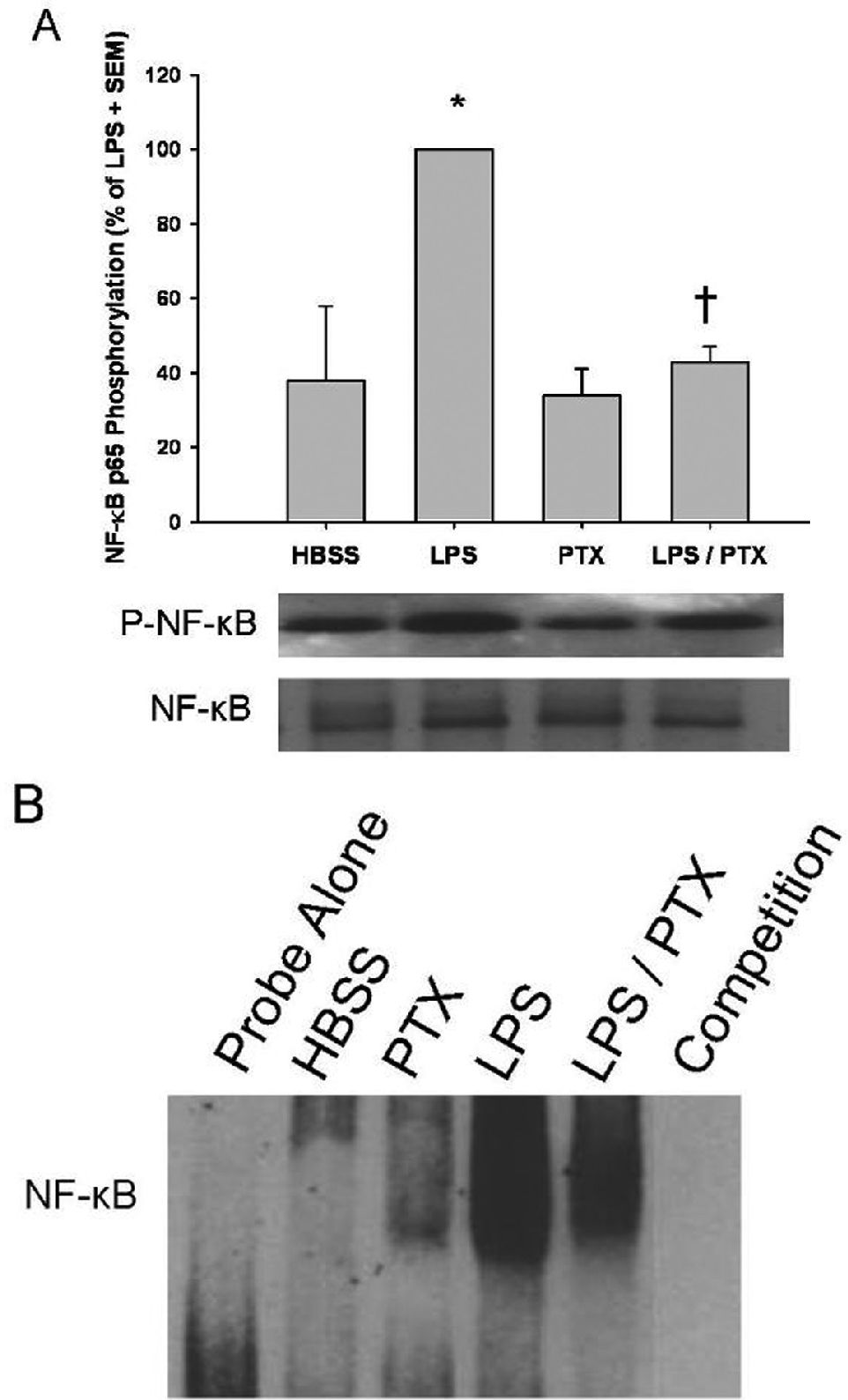
In a similar fashion, nuclear NF-κB p65 phosphorylation was increased following LPS stimulation (100 ± 0 vs. 38 ± 20; P < 0.01). PTX similarly decreased LPS-induced NF-κB p65 nuclear phosphorylation, a marker of NF-κB activation and nuclear translocation (100 ± 0 vs. 40 ± 6; P =0.03; Figure 3A).
 were incubated with LPS (1 μg/mL) in the presence and absence Pentoxifylline (PTX; 20 mM). A, Nuclear NF-κB p65 phosphorylation was assessed by western blotting, and results are expressed as a percentage of LPS stimulation ± SEM (n = 4 per group). LPS-stimulation markedly enhanced NF-κB phosphorylation over that of the control (*, P < 0.01 vs. HBSS). The addition of PTX resulted in diminished NF-κB phosphorylation (†, P = 0.03 vs. LPS). B, NF-κB DNA-binding was determined by EMSA, as described in Materials and Methods. NF-κB-DNA binding was upregulated following LPS treatment and attenuated following the addition of PTX in a manner that was similar to NF-κB p65 subunit phosphorylation.")
Effects of phosphodiesterase inhibition on LPS-induced NF-κB phosphorylation. Isolated human mononuclear cells (5 x 106) were incubated with LPS (1 μg/mL) in the presence and absence Pentoxifylline (PTX; 20 mM). A, Nuclear NF-κB p65 phosphorylation was assessed by western blotting, and results are expressed as a percentage of LPS stimulation ± SEM (n = 4 per group). LPS-stimulation markedly enhanced NF-κB phosphorylation over that of the control (*, P < 0.01 vs. HBSS). The addition of PTX resulted in diminished NF-κB phosphorylation (†, P = 0.03 vs. LPS). B, NF-κB DNA-binding was determined by EMSA, as described in Materials and Methods. NF-κB-DNA binding was upregulated following LPS treatment and attenuated following the addition of PTX in a manner that was similar to NF-κB p65 subunit phosphorylation.
EMSAs were then performed to verify the PTX-induced alterations in nuclear phosphorylation resulted in similar effects on the DNA binding activity of NF-κB. The addition of PTX to LPS-stimulated mononuclear cells resulted in comparable downregulation of DNA binding activity, similar to the observed downregulation of NF-κB phosphorylation (Figure 3B).
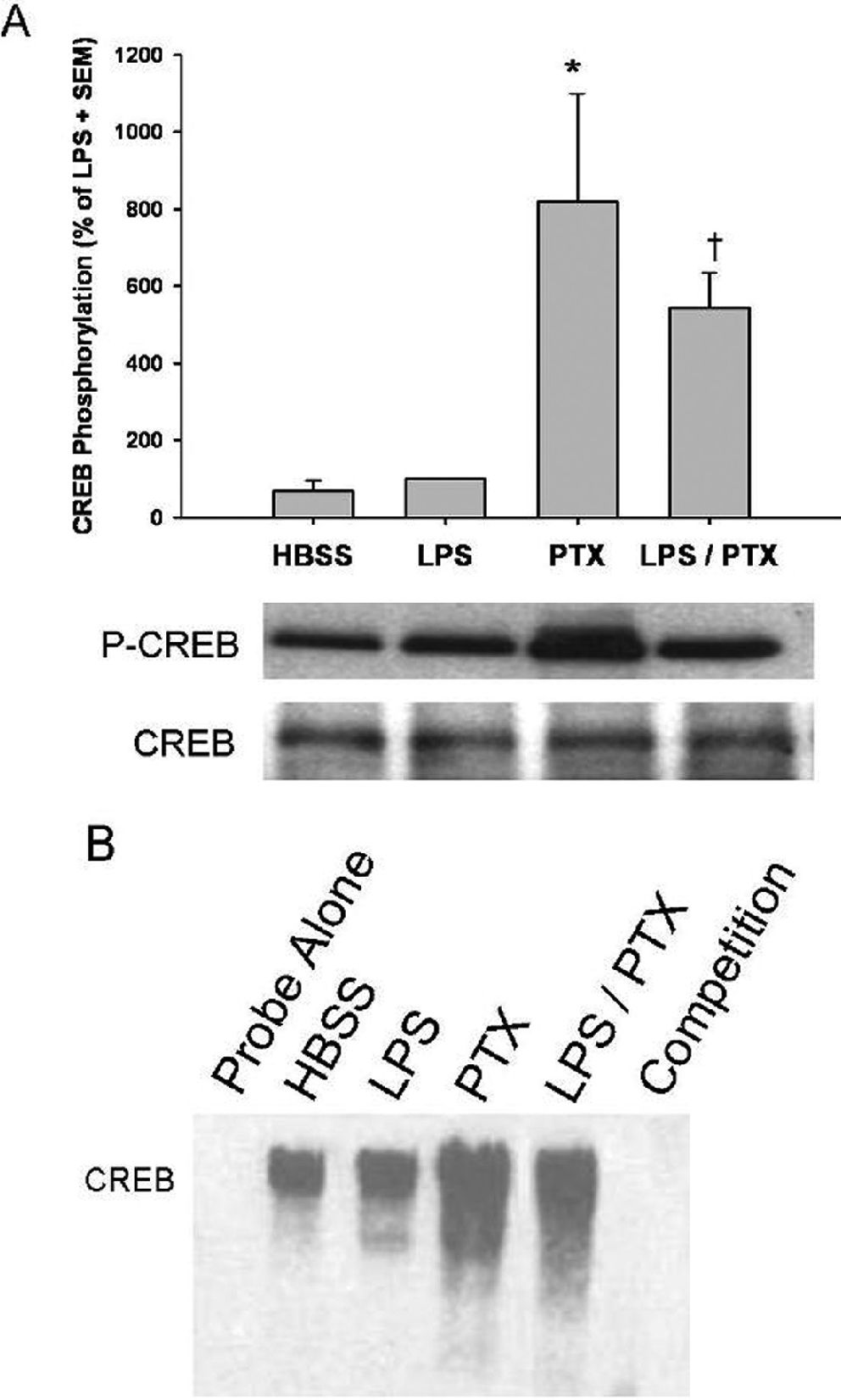
PTX upregulates CREB phosphorylation and activation after LPS stimulationPhosphorylation of nuclear CREB was used as a marker for CREB activation. LPS stimulation caused a negligible increase in CREB phosphorylation when compared to control (Figure 4A). PTX alone caused a marked increase in CREB phosphorylation (P < 0.01 vs. HBSS). When LPS and PTX exposure occurred simultaneously, CREB phosphorylation was significantly higher than with LPS stimulation alone (543 ± 92 vs. 100 ± 0; P < 0.01). In addition, the amount of CREB phosphorylation seen with concomitant LPS and PTX treatment was less than that seen with PTX alone, although this difference was not statistically significant (P = 0.08).
 were incubated with LPS (1 μg/mL) in the presence and absence Pentoxifylline (PTX; 20 mM). A, CREB phosphorylation was assessed by western blotting, and the results are expressed as a percentage of LPS stimulation ± SEM (n = 4 per group). Incubation of cells with PTX alone resulted in a concentration-dependent upregulation of CREB phosphorylation (*, P < 0.05 vs. HBSS). The concomitant administration of LPS slightly attenuated phosphorylation, which nonetheless remained significant when compared to that observed in cells treated with LPS alone (†, P < 0.01 vs. LPS). B, CREB-DNA binding was determined by EMSA, as described in Materials and Methods. CREB-DNA binding after LPS stimulation was similar to that of the control. PTX upregulated CREB-DNA binding in the presence and absence of LPS in a manner that was similar to CREB phosphorylation.")
Effects of phosphodiesterase inhibition on LPS-induced CREB phosphorylation. Isolated human mononuclear cells (5 x 106) were incubated with LPS (1 μg/mL) in the presence and absence Pentoxifylline (PTX; 20 mM). A, CREB phosphorylation was assessed by western blotting, and the results are expressed as a percentage of LPS stimulation ± SEM (n = 4 per group). Incubation of cells with PTX alone resulted in a concentration-dependent upregulation of CREB phosphorylation (*, P < 0.05 vs. HBSS). The concomitant administration of LPS slightly attenuated phosphorylation, which nonetheless remained significant when compared to that observed in cells treated with LPS alone (†, P < 0.01 vs. LPS). B, CREB-DNA binding was determined by EMSA, as described in Materials and Methods. CREB-DNA binding after LPS stimulation was similar to that of the control. PTX upregulated CREB-DNA binding in the presence and absence of LPS in a manner that was similar to CREB phosphorylation.
To determine if CREB-DNA binding was affected by PTX in a manner similar to CREB phosphorylation, an EMSA was performed. Exposure of LPS-stimulated cells to PTX similarly increased CREB DNA-binding compared to LPS alone (Figure 4B).
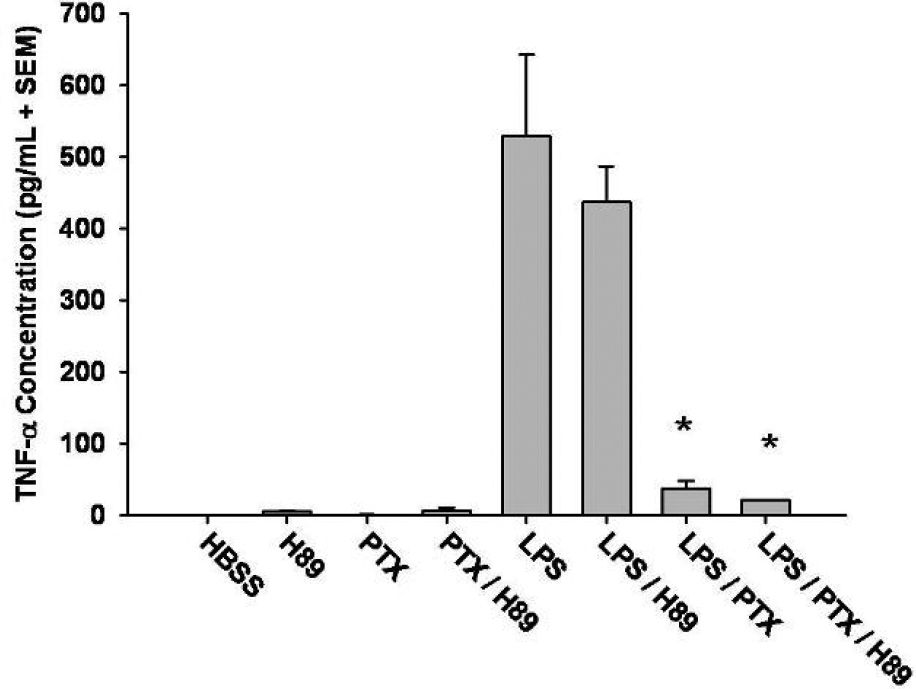
Effect of PTX on TNF-α production in the presence of PKA inhibitionThe role of PKA in PTX-induced downregulation of TNF-α production in LPS-stimulated mononuclear cells was assessed by treating mononuclear cells with H89, a PKA inhibitor, prior to LPS and PTX exposure. The addition PTX to LPS-stimulated cells resulted in a reduction in TNF-α, similar to that observed in our previous experiments (529 pg/mL ± 112 vs. 37 pg/mL ± 10; P < 0.01). Pre-incubation of cells with H89 prior to LPS and PTX exposure did not result in any significant modulation of TNF-α production when compared to that demonstrated with PTX alone (P = 0.2; Figure 5). These results suggest that downregulation of TNF-α production by PTX in LPS-stimulated mononuclear cells is, at least in part, PKA-independent.
. Isolated mononuclear cells (5 x 106) were preincubated with the PKA inhibitor, H89 (10 μM), for one hour prior to stimulation with LPS (1 μg/mL) or LPS (1 μg/mL) and Pentoxifylline (PTX; 20 mM) for an additional 30 minutes. The concentration of TNF-α present in the supernatant was measured by ELISA. PTX downregulated LPS-induced TNF-α expression (*, P < 0.01 vs. LPS). The addition of H89 did not significantly alter the effects of PTX on TNF-α production after LPS stimulation.")
Phosphodiesterase inhibition downregulates LPS-induced TNF-α production independent of Protein kinase A (PKA). Isolated mononuclear cells (5 x 106) were preincubated with the PKA inhibitor, H89 (10 μM), for one hour prior to stimulation with LPS (1 μg/mL) or LPS (1 μg/mL) and Pentoxifylline (PTX; 20 mM) for an additional 30 minutes. The concentration of TNF-α present in the supernatant was measured by ELISA. PTX downregulated LPS-induced TNF-α expression (*, P < 0.01 vs. LPS). The addition of H89 did not significantly alter the effects of PTX on TNF-α production after LPS stimulation.
The exposure of inflammatory cells to a variety of extracellular stimuli results in the initiation of transcriptional and posttranscriptional events, which culminate in the production of pro-inflammatory mediators.11–14 TNF-α is released very early following shock and is considered an important mediator of the inflammatory cascade, since it regulates the synthesis of several other critical cytokines and chemokines.15 A clear relationship between increased TNF-α synthesis and the development of shock and multiple organ dysfunction has been observed in animal models of sepsis.15–17
Because of its importance in orchestrating multiple steps of the inflammatory response, strategies aimed at decreasing TNF-α levels or attenuating its production seem attractive. PDE inhibitors have been shown to downregulate TNF-α production in distinct human cell populations. We have previously shown that, in general, TNF-α levels continue to increase after LPS exposure and that concomitant infusion of phosphodiesterase inhibitors can markedly decrease TNF-α levels.18–19 In this in vitro study, we verified the sustained attenuation of LPS-induced TNF-α expression over time in an isolated human mononuclear cell population.
The mechanisms by which PTX, a non-specific phosphodiesterase inhibitor, significantly and consistently decreases TNF-α production have not been completely elucidated. It has been postulated that drugs that increase intracellular cAMP exert their anti-inflammatory effects through the activation of PKA. This classic pathway has been challenged as the sole mechanism involved in the modulation of inflammation by cAMP-enhancing drugs, and alternative mechanisms involving PKA-independent pathways have been proposed.6–8 In this series of experiments, we observed a significant attenuation of TNF-α production in LPS-stimulated human mononuclear cells following PTX administration, independent of PKA activation.
We used a nonspecific phosphodiesterase inhibitor, PTX, in the present study because it has been used clinically in the treatment of a variety of conditions in which inflammation is an important component of the pathophysiology of the disease process.19 Additionally, the use of PTX in sepsis as an adjunct to other treatments to maintain adequate organ function has been explored based upon its effects on TNF-α synthesis. However, the reported beneficial effects of PTX are not only related to the downregulation of TNF-αbut also to PTX’s hemorrheologic properties, its ability to reduce neutrophil activation, and its beneficial effects on microcirculation, cardiac performance, and organ injury.20–25 Herein, we demonstrated that the downregulatory effects of PTX in human mononuclear cells involves modulation of pathways that involve NF-κB and CREB, two major transcription factors involved in the inflammatory cascade.
NF-κB/Rel constitutes a family of transcriptional factors involved in the regulation of numerous cytokine genes and immune responses in different cell populations. The most abundant form of NF-κB is the p50–p65 heterodimer.26 The inactive form of NF-κB exists in the cytoplasm bound to an inhibitory complex containing I-κBα. Following cellular stimulation with LPS and chemotactic factors, NF-κB inducible kinase phosphorylates and activates the I-κB kinase (IKK) complex consisting of IKK-1 and IKK-2, which in turn phosphorylates the I-κBα subunit. Phosphorylation, ubiquitination, and subsequent degradation of I-κBα results in NF-κB nuclear translocation. This allows the transcription factor to bind to promoter regions of specific pro-inflammatory genes and influence transcription.27,28 Because a number of potential κB sites are present in the nucleotide sequences of the TNF-α gene, NF-κB is considered to be a critical transcriptional factor involved in TNF-α gene expression and protein synthesis.29,30
In this set of experiments, we demonstrated that PTX downregulates cytoplasmic I-κBα phosphorylation, nuclear NF-κB p65 phosphorylation/translocation, as well as NF-κB DNA-binding after LPS stimulation, suggesting that PTX exerts its function proximal to or at the level of I-κBα phosphorylation.
Previous studies that have evaluated the effects of cAMP-elevating drugs on I-κBα and NF-κB have reported conflicting results. Haddad et al. reported results similar to ours with respect to the effects of PTX on I-κBα and NF-κB in pulmonary epithelial cells.31 Conversely, Neumann et al. proposed that increased intracellular cAMP stabilizes the interaction between I-κBα and NF-κB, therefore reducing NF-κB activation.32 In contrast, Takahashi et al. showed no difference in I-κBα degradation in Jurkat T-lymphocytes in the presence and absence of forskolin, an alternative cAMP inducing compound. This group also demonstrated that forskolin did not inhibit the DNA-binding activity of NF-κB and that CREB was not involved in forskolin-induced decrease in p65 transcriptional activity.33 Similar results were reported by Ollivier et al. using THP-1 cells and endothelial cells treated with forskolin or dibutyryl cAMP, two agents known to function through cAMP elevation and subsequent PKA activation.9 The differences between our results and those mentioned above may be explained by the use of different cell types and exogenous stimulants. It is also possible that PTX exerts its anti-inflammatory effects on the NF-κB pathway independent of PKA, as suggested by the continued attenuation of LPS-induced TNF-α production by PTX in the presence or absence of PKA inhibition observed in this study. This hypothesis is currently under investigation in our laboratory. Furthermore, we utilized a broad PKA inhibitor in this study in addition to H89, but did not examine the effect of specific inhibition of various PKA isoforms. This information may yield more insight into the mechanism by which PTX exerts its down-regulatory effects.
PDE inhibitors may also downregulate NF-κB transcriptional activity by altering the competitive binding of CREB and NF-κB to the promoter regions of pro-inflammatory genes. Herein, we showed that the activation and DNA binding activity of CREB is upregulated in a dose–dependent manner by PTX. Of note, the increase in CREB activity was slightly alleviated by the addition of LPS, suggesting competition between PTX-induced CREB-associated gene transcription and those factors, such as NF-κB, which may be upregulated by LPS exposure. This competition has been postulated to involve the recruitment of the co-activator CBP and its homologue p30034, suggesting an alternative mechanism that may be involved in the anti-inflammatory actions of PTX. Future studies are planned to delineate the validity of this potential mechanism.
The effects of PTX on other transcription factors, such as AP-1, c-fos, and c-jun, cannot be ruled out by our experiments. Activation of AP-1 and its components (c-fos and c-jun) by agents that enhance mononuclear cell activity has been implicated in TNF-α expression. Moreover, AP-1 and CREB recognize a similar DNA binding sequence in the promoter region of the TNF-α gene35. Given these relationships, it is conceivable that PTX may exert some of its anti-inflammatory actions by affecting AP-1 activation and favoring CREB binding, thus inhibiting TNF-α gene transcription. Additional studies are necessary to elucidate the complex interactions between CREB, NF-κB, and AP-1 following LPS exposure and PDE inhibition. Furthermore, the effect of PTX on the cGMP pathway after LPS stimulation has not been explored, although we plan to do so in the future.
In summary, we have demonstrated that PDE inhibition of human mononuclear cells downregulates TNF-α production, at least in part, through a PKA-independent mechanism. In addition, PTX attenuates the activity of NF-B while upregulating CREB activation after LPS stimulation, which may result in modulation of pro-inflammatory mediator synthesis. Therefore, PTX may serve as a potential adjunct therapeutic for the treatment of conditions in which TNF-α production plays a significant role.