




Cellular energy metabolism is one of the main processes affected during the transition from normal to cancer cells, and it is a crucial determinant of cell proliferation or cell death. As a support for rapid proliferation, cancer cells choose to use glycolysis even in the presence of oxygen (Warburg effect) to fuel macromolecules for the synthesis of nucleotides, fatty acids, and amino acids for the accelerated mitosis, rather than fuel the tricarboxylic acid cycle and oxidative phosphorylation. Mitochondria biogenesis is also reprogrammed in cancer cells, and the destiny of those cells is determined by the balance between energy and macromolecule supplies, and the efficiency of buffering of the cumulative radical oxygen species. In glioblastoma, the most frequent and malignant adult brain tumor, a metabolic shift toward aerobic glycolysis is observed, with regulation by well known genes as integrants of oncogenic pathways such as phosphoinositide 3-kinase/protein kinase, MYC, and hypoxia regulated gene as hypoxia induced factor 1. The expression profile of a set of genes coding for glycolysis and the tricarboxylic acid cycle in glioblastoma cases confirms this metabolic switch. An understanding of how the main metabolic pathways are modified by cancer cells and the interactions between oncogenes and tumor suppressor genes with these pathways may enlighten new strategies in cancer therapy. In the present review, the main metabolic pathways are compared in normal and cancer cells, and key regulations by the main oncogenes and tumor suppressor genes are discussed. Potential therapeutic targets of the cancer energetic metabolism are enumerated, highlighting the astrocytomas, the most common brain cancer.
The incidence of primary brain tumors is estimated at 7.2–12.5 per 100 million persons per year, representing around 2% of all adult primary tumors and 23% of cancer in childhood. Mortality resulting from primary brain tumor amounts to 13,000 deaths per year, which is 2% of adult and 26% of childhood cancer deaths.1–4 Astrocytoma, a brain tumor originating in glial cell types, is the most frequent brain tumor, and glioblastoma (GBM), the grade IV astrocytoma, is the most malignant and frequent of these.5 In spite of the introduction of new molecular-based therapies, this mortality has not changed much during the last three decades.6–9 The survival outcome for GBM patients has improved from 3 months following surgical resection only to 8 months with the introduction of radiotherapy10 and to 6.9 months of median progression-free survival and median overall survival of 14.6 months with the further addition of temozolomide concurrently with irradiation.11 Diffuse infiltration of tumor cells into normal brain tissue presents difficulties in surgical resection and partially explains the poor outcome. Furthermore, the diverse causative genotypes leading to a heterogeneous histological phenotype are additional characteristics of this tumor offering obstacles to effective therapy. Recent studies demonstrating the presence of multiple mutations in brain tumors, specifically in GBM12 and medulloblastoma,13 corroborate the hypothesis that the development of a brain tumor also requires the acquisition of several mutations, as described previously for colorectal carcinoma.14 At the same time, the Cancer Genome Atlas Research Network performed a large-scale multi-dimensional analysis of the molecular characteristics of GBM and provided a network view of the pathways altered in the development of GBM.15 Nonetheless, cumulative evidence shows that mutations are not the only cause of the altered gene expression of cancer cells, and that epigenetic alterations,16,17 heritable and reversible changes other than the DNA sequences,18 and aneuploidy, numerical and structural abnormalities in chromosomes,19 are common alterations in tumor cells and may also modify gene expression and play a crucial role in tumorigenesis. Epigenetic modulation of gene expression is essential for normal cellular development, and promoter CpG island hypermethylation and transcriptional silencing of tumor suppressor genes and pro-differentiation factors are hallmarks of epigenetic alteration in cancer cells. For example, methylation of the O-6-methylguanine-DNA methyltransferase promoter in GBM has been shown to be a useful predictor of the responsiveness of the tumors to alkylating agents.20–22 In addition, cellular energy metabolism is one of the main processes affected during the transition from normal to cancer cells. Metabolic activity is a relevant determinant of a cell decision to proliferate or die. Cancer cells alter their metabolism in order to support rapid proliferation. Otto Warburg, Nobel Prize laureate for Physiology Medicine in 1931, demonstrated that cancer cells do not metabolize glucose in the same way as normal adult differentiated cells. Cancer cells generally use glycolysis even in the presence of abundant oxygen, a phenomenon named aerobic glycolysis, the Warburg effect,23,24 rather than fuel the tricarboxylic acid (TCA) cycle. It is currently believed that the glycolytic switch is acquired very early in tumorigenesis even before tumors experience hypoxia.25
In the present review, the main metabolic pathways in normal adult cells are described first, then the modifications that occur in cancer cells are presented highlighting the potential therapeutic targets, and finally, the interactions between oncogenes and tumor suppressor genes with metabolic pathways are discussed.
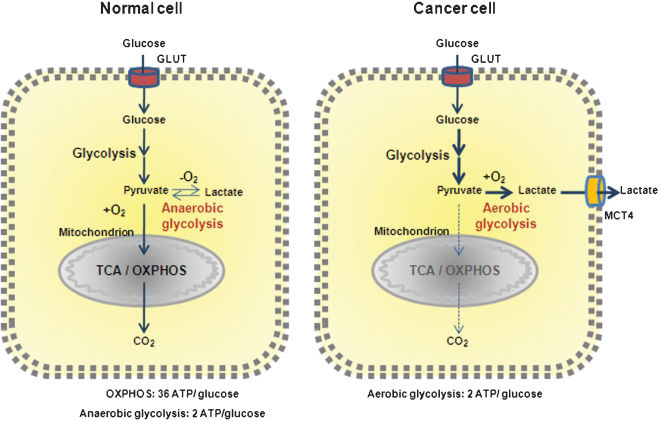
Metabolism in normal cellsIn normal cells, to produce two viable daughter cells at mitosis, all the cellular contents must be replicated, and energy is necessary for this to happen. Glucose participates in cellular energy production with two adenosine triphosphate (ATP) synthesis through glycolysis and up to 36 ATPs through its complete catabolism by the TCA cycle and OXPHOS (oxidative phosphorylation) (Figure 1). The large requirements for nucleotides, amino acids, and lipids for the daughter cells are provided by intermediate metabolites of these pathways. In addition to glucose, glutamine is the other molecule catabolized in appreciable quantities for most mammalian cells in culture. Both molecules supply carbon, nitrogen, free energy, and reducing equivalents necessary to support cell growth and division. This means that glucose, in addition to being used for ATP synthesis, should also be diverted to macromolecular precursors such as acetyl-CoA for fatty acids, glycolytic intermediates for non-essential amino acids, and ribose for nucleotides to generate biomass.
, diverting glucose metabolites from energy production to anabolic process to accelerate cell proliferation, at the expense of generating only two ATPs per glucose.")
Metabolic differences between normal and cancer cells. Normal cells primarily metabolize glucose to pyruvate for growth and survival, followed by complete oxidation of pyruvate to CO2 through the TCA cycle and the OXPHOS process in the mitochondria, generating 36 ATPs per glucose. O2 is essential once it is required as the final acceptor of electrons. When O2 is limited, pyruvate is metabolized to lactate. Cancer cells convert most glucose to lactate regardless of the availability of O2 (the Warburg effect), diverting glucose metabolites from energy production to anabolic process to accelerate cell proliferation, at the expense of generating only two ATPs per glucose.
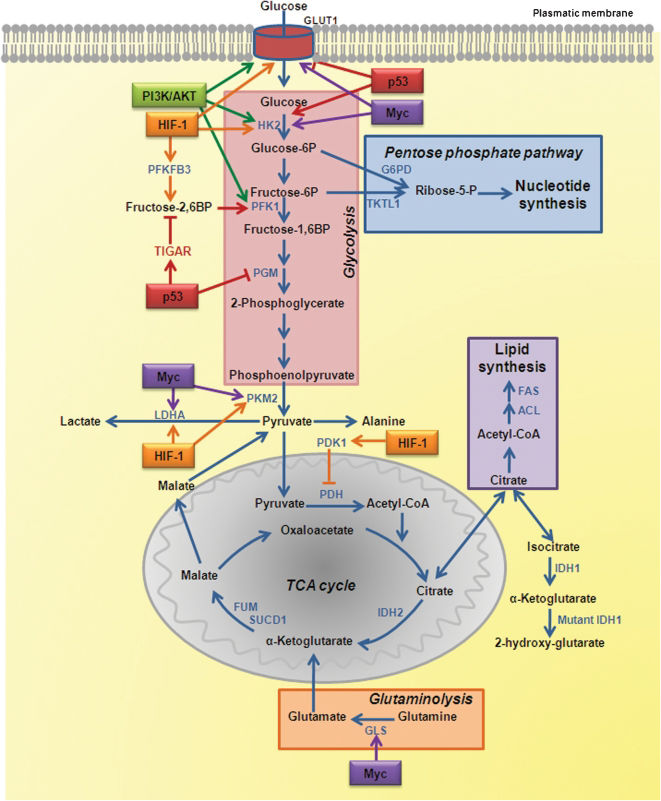
Glucose enters the cell through glucose transporters (GLUTs) and, once intracellular, is phosphorylated to glucose-6-phosphate (G6P) by hexokinase 2 (HK2). Phosphoglucose isomerase catalyzes G6P to fructose-6-phosphate (F6P), which yields fructose-1,6-biphosphate by phosphofructokinase 1 (PFK1), and then pyruvate and ATP by pyruvate kinase (PK) in the final step of glycolysis. Pyruvate is converted to acetyl-CoA, which enters the TCA cycle. Ultimately, glycolysis produces two ATP molecules and six NADH molecules per glucose. In normal tissues, most of the pyruvate is directed into the mitochondrion to be converted into acetyl-CoA by the action of pyruvate dehydrogenase (PDH) or transaminated to form alanine (Figure 2).

Metabolic remodeling in cancer cells and regulation by signaling pathways involving oncogenes and tumor suppressor genes. The key enzymes of glycolysis, the TCA cycle, the pentose phosphate pathway, glutaminolysis, nucleotide, and lipid biosynthesis are shown as the regulation points by oncogenes and tumor suppressor genes.
This is a metabolic pathway that generates NADP and pentose sugars from G6P. The enzyme that governs the entry of G6P into this pathway is glucose-6-phosphate dehydrogenase (G6PD), which is regulated by the availability of its substrate and the NADPH to NADP+ ratio.26 G6P is converted to ribose-5-phosphate (R5P) while producing two molecules of NADPH. NADPH is both a major cellular antioxidant, maintaining glutathione in a reduced state to prevent oxidative damage, and a required cofactor in the reductive biosynthesis of fatty acids, nucleotides, and amino acids. NADH is also used in mitochondrial OXPHOS (Figure 2).
Tricarboxylic acid (TCA) cyclePyruvate produced by glycolysis is converted to acetyl-CoA, which enters the TCA cycle, and citrate, α-ketoglutarate, succinyl-CoA, fumarate, malate, and oxaloacetate are produced as intermediate products. Most of the carbon for fatty acids derives from acetyl-CoA synthesized in the mitochondrial matrix. However, acetyl-CoA cannot cross the inner mitochondrial membrane, but intramitochondrial acetyl-CoA and oxaloacetate combine to form citrate, which is transported out of the mitochondria and broken back down into its constituents by ATP citrate lyase (ACL). Acetyl-CoA is converted to malonyl-CoA by acetyl-CoA carboxylase, and acetyl-CoA and malonyl-CoA are then both used by the multi-subunit enzyme fatty acid synthase (FAS) for the synthesis and elongation of fatty acid chains. Oxaloacetate is used for the synthesis of non-essential amino acids. Cytosolic and nuclear acetyl-CoA is also a precursor for the post-translational modification of proteins (for example, histones) by acetylation. Similarly to citrate, malate produced in the TCA cycle leaves the mitochondria, and it is converted to pyruvate plus NADPH. Citrate might also be converted to isocitrate and then to α-ketoglutarate, generating another molecule of NADPH by the action of isocitrate dehydrogenase 1 (IDH1). In addition to glucose, amino acids can also fuel the TCA cycle. Glutamine supplies carbon in the form of mitochondrial oxaloacetate to maintain citrate production in the first step of the TCA cycle (Figure 2).
GlutaminolysisGlutamine contributes both to the substrate needs of a dividing cell and to the control of redox potentials through the synthesis of NADH. After glutamine is taken into the cell, a mitochondrial-associated enzyme, glutaminase-1 (GLS), converts it to glutamate. Glutamate is converted to α-ketoglutarate and enters the TCA cycle in the mitochondria (Figure 2). Glutamate can also be converted to aspartate, which contributes to nucleotide synthesis. The excessive quantity of glutamine used by the cells results in alanine and ammonium secretions.
Metabolic modifications in cancer cells and potential targets for therapyCancer cells adapt themselves to maximize their ability to synthesize substrates for membranes, nucleic acids, and proteins for the increased proliferative rate, a major characteristic of these cells. This cannot be accomplished without large amounts of energy (ATP), which are obtained by increasing the use of glucose and glutamine many times. The cancer cells rely on aerobic glycolysis, the Warburg effect,23,24 with a reduced use of the TCA cycle, so that the pyruvate made in glycolysis is converted to lactate27 (Figure 1). In fact, the fate of pyruvate depends on many factors, of which oxygen availability is one of the most important. In the presence of oxygen, the pyruvate is directed into the mitochondrion to be converted into acetyl-CoA by the action of PDH or transaminated to form alanine. And then, once inside the mitochondrion, pyruvate is completely oxidized through the TCA cycle and OXPHOS. However, in cancer cells, PDH activity is blocked by the hypoxia-driven enzyme pyruvate dehydrogenase kinase 1 (PDK1).28,29 In addition, an increase in lactate dehydrogenase A (LDHA) enzymatic activity is observed in cancer cells (Figure 2).30,31 These two facts determine the fate of pyruvate, which is converted into lactate, contributing to the Warburg effect and to the enhancement of the malignant phenotype.32,33 Animal experiments knocking down LHDA or inhibiting PDK1, using interference RNA or dichloroacetate, have demonstrated a reduction in tumor growth in xenograft models.30,31,34 Such results corroborate the importance of the decrease in the rate of pyruvate entering the TCA cycle and the concurrent increase in lactate production for the growth and survival of tumor cells. The lactate produced is exported from the cells by monocarboxylate transporter 4 (MCT4), which allows the cell to preserve normal cellular pH (Figure 1). Oxygenated normal cells can remove lactate from the extracellular fluid using monocarboxylate transporter 1, and convert it back to pyruvate for further oxidation, using the lactate dehydrogenase B (LDHB) isoform. However, the same is not observed in hypoxic cancer cells, and the large amounts of exported lactate create an acidic tumor environment, which encourages cancer cell invasion. Thus, therapies targeting tumor acidification would inhibit glycolytic energy production and may also inhibit tumor cell invasion.35 Nevertheless, treatment regimens directed toward such ubiquitous transporters, such as monocarboxylate transporters, are also likely to affect normal tissues. Therefore, either their action must be extremely rapid or the dose very low to avoid side-effects in other tissues.
The pyruvate synthesis itself by PK is also an important energy-producing step in glycolysis. This step is highly regulated by isoform selection and by allosteric regulation. Four PK isoforms have been described in humans: PKM1, PKM2, PKL, and PKR. The PKM1 isoform is expressed only in normal tissues and is incompatible with tumor growth. This isoform is replaced by the alternative spliced form PKM2 in highly proliferative tumor cells.36,37 PKL is found in the liver and kidney, and PKR in erythrocytes. PKM2 oscillates between inactive dimeric and active tetrameric forms. The formation of PKM2 tetramers is stimulated by the glycolytic intermediate fructose-1,6-biphosphate.38 The dimeric form of PKM2 retards pyruvate formation and allows the accumulation of upstream glycolytic intermediates, promoting their distribution into the biosynthetic pathways. The tetrameric form of PKM2 channels pyruvate to lactate. Phosphotyrosine residues bind to the same allosteric regulatory site of fructose-1,6-biphosphate, releasing it.39,40 This regulation of enzyme activity may constitute a molecular switch that diverts glucose metabolites from energy production to anabolic processes when cells are stimulated by certain tyrosine phosphorylated growth factors. If an inhibitor could be designed for the tumor-expressed PKM2 isoform, it might specifically inhibit glycolysis in tumor cells, killing them by energy deficit. A therapeutic strategy inhibiting the interaction between phosphotyrosine residues and PKM2 to block the anabolic process of tumor cells would also be an interesting avenue to explore.
PFK1 is also a rate-limiting enzyme in glycolysis41 and highly activated in tumor cells. PFK1 adds a second phosphate group to F6P. One potent allosteric activator of PFK1 is fructose-2,6-biphosphate (F2,6BP), the product of the 6-phosphofructose-2-kinase/fructose2,6-biphosphatase (PFKFB). PFKFBs are bifunctional enzymes that interconvert F6P and F2,6BP. PFK1 is normally inhibited by ATP, but F2,6BP overrides this inhibition and enhances the glycolytic flux, allowing tumor cells to maintain high glycolytic flux despite the presence of physiological levels of ATP. An isoform of PFKFB, isoenzyme 3 (PFKFB3), was recently shown to promote proliferation through its effects on cell cycle regulators. PFKFB3 is upregulated in tumors, and its inhibition has been proved to decrease F2,6BP levels, which in turn decreases PFK1 activity and glycolytic flux with cytostatic effect (Figure 2).42
HK2 participates in the glycolytic pathway and also in metabolite transport into and out of the mitochondrial intermembrane space through its association with a voltage-dependent anion channel,43 a 30 kDa pore protein inserted in the outer mitochondrial membrane.44 Therefore, HK2 is another potential therapeutic target. In fact, two hexokinase inhibitors, lonidamine and glucose mimetic, 2-deoxyglucose, are currently in clinical trials in combination with other agents.45 3-Bromopyruvate is a further inhibitor of HK2 with promising in vivo studies, but there is no current clinical trial.46,47
The modifications in cancer cells enumerated above permit an understanding of how these cells are urged to a non-profitable choice of two ATP generation by aerobic glycolysis instead of 36 ATP generation upon complete glucose oxidation by the TCA cycle and OXPHOS. In spite of this apparently “bad” choice, cancer cells continue to exhibit high ratios of ATP/adenosine diphosphate (ADP) and NADH/NAD+48 due to an alternative ATP production by converting two ADPs to one ATP and one adenosine monophosphate (AMP) catalyzed by adenylate kinases. This not only helps to maintain a viable ATP/ADP ratio as ATP production declines, but also to accumulate AMP, which activates AMP-kinase and leads to the phosphorylation of several targets to improve energy charge in cells.49
Another important reason for the cancer cells to switch to aerobic glycolysis is to provide metabolic macromolecules for the daughter cells. 13C-nuclear magnetic resonance spectroscopy measurements show that 90% of glucose and 60% of glutamine are converted into lactate or alanine by GBM cell cultures.50 Although each lactate excreted from the cell wastes three carbons that might otherwise be utilized for either ATP production or macromolecular precursor biosynthesis, the tumor cells choose this method to fasten carbon incorporation into biomass to increment cell division velocity. Glutaminolysis also generates reductive power required for fatty acid biosynthesis by NADPH production via the activity of NADP+-specific malate dehydrogenase (malic enzyme), in addition to the fundamental role in replenishing the TCA cycle.51 Blocking the fuel through this pathway for the biomass to tumor proliferation seems a good therapeutic strategy. Phenylacetate is a drug that reduces the biological availability of glutamine in the blood. This reagent condenses with the γ-amino group of glutamine and is excreted into urine. A previous report has demonstrated that phenylacetate inhibits the proliferation of glioma cells and promotes their differentiation.52 However, the removal of glutamine directly from the plasma may also increase the rate at which the body cannibalizes its own muscles (cachexia). Additionally, various other anti-glutaminolysis compounds have been developed, but they were found to be toxic or raised immune reactions.53
Cancer biomass reduction may also be achieved by blocking fatty acid synthesis through the inhibition of ACL, which converts acetyl-CoA to malonyl-CoA,54 and FAS, a multifunctional protein that converts malonyl-CoA to palmitate over multiple steps.55 Inhibition of both enzymes, ACL and FAS, has been shown to limit tumor cell proliferation and survival in vitro and in vivo.55,56 One possible negative aspect of anti-FAS therapy is its effect on food intake and body weight, as observed in treated rodents that presented hypophagia and consequent weight loss.57 Deficiencies in two other enzymes participating in the TCA cycle, fumarate hydratase (FUM) and succinate dehydrogenase 1 (SUCD1), may also have a tumor suppressive effect (Figure 2).58
Nucleotide biosynthesis has also been targeted to block biomass production in cancer cells for several years. 5-fluorouracil, cytarabine, and methotrexate are examples of chemotherapeutic agents known as antimetabolites. Most of these drugs target the final stages in the nucleotide synthetic pathway, and therefore lack specificity, leading to nucleotide shortage, incomplete DNA synthesis, and cell death indistinctly of tumor and normal proliferating cells.59 Therefore, blocking the early stages of nucleotide biosynthesis such as R5P production could provide a better therapeutic window than that shown by previous antimetabolic therapies. Transketolase-like protein 1 (TKTL1), an enzyme in the non-oxidative arm of the PPP that produces R5P, has been found to be upregulated in several tumor types,60 and knocking it down reduced the proliferation of tumor cells, as well as decreasing lactate production and resensitizing cells to reactive oxygen species (ROS)-generating compounds.61 Isolated mutations in either TKTL1 or G6PD of PPP have no impact on cancer cell growth, as both enzymes contribute to R5P production. However, simultaneous mutations in these two genes, blocking R5P synthesis, are lethal for cancer cells in animals (Figure 2).62
Besides the alterations described above in the main metabolic routes, side pathways of the TCA cycle are also implicated in cancer progression. A highly prevalent mutation in an enzyme related to metabolism was uncovered in the recent high throughput mutation screening in GBM, which highlights the importance of energetic metabolism in tumor progression. This study revealed that up to 12% of the GBMs harbor the same mutation in the gene encoding cytosolic IDH1.12,63 Monoallelic mutation in the same residue 132 in IDH1 (IDH1R132) or the analogous residue in the related enzyme IDH2 is a common feature of gliomas, as more than 80% of indolent gliomas harbor such a mutation.64,65 IDH1 and IDH2 couple the reversible conversion of isocitrate to α-ketoglutarate and NADP+ to NADPH. IDH1 is located in the cytosol and the peroxisome66,67 and produces NADPH.68 In the peroxisome, NADPH contributes to cholesterol synthesis.69 IDH2 is located in the mitochondria and catalyzes the isocitrate to α-ketoglutarate reaction in the TCA cycle.70 The occurrence of IDH1 mutations correlated with approximately twofold diminished NADP+-dependent IDH activity, and total NADPH production is hampered by 38% in GBM harboring the IDH1R132 mutation. Therefore, mutated IDH1 consumes rather than produces NADPH. NADPH/NADH is both a major antioxidant, maintaining glutathione in a reduced state, protecting the cell from ROS, and a required cofactor in the biosynthesis of fatty acids, nucleotides, and amino acids. Thus, the NADPH level may affect not only cellular proliferation but also mutation rates.71 Then, the low NADPH levels due to IDH1R132 mutation in GBM may sensitize tumors to irradiation and chemotherapy.72 In contrast, IDH1R132 mutation also leads to the accumulation of 2-hydroxyglutarate which, based on its structural similarity to α-ketoglutarate, may competitively inhibit prolyl hydroxylase, which targets hypoxia induced factor 1-α (HIF-1α) for ubiquitylation and subsequent proteasomal degradation. Therefore, the substrate of the IDH1R132 mutation stabilizes HIF-1α, which activates metabolic changes, as described below, and stimulates invasion, cell survival, and angiogenesis (Figure 2).73,74
An important penalty for this increased flux of macromolecules to provide biomass for the proliferating cancer cells not converted to aerobic glycolysis is also an increase in mitochondrial OXPHOS, the major source of ROS production. Elevated mitochondrial ROS formation frequently occurs upon suppression of pyruvate input into OXPHOS.75 This is justified only when additional blockage exists within the electron transport chain, such as that caused by mutations or an insufficiency of mitochondria-encoded subunits, or HIF-1 and oncogene-mediated suppression of electron transport chain components. Slow electron transport at a relatively high substrate also pressures the electron transport chain with a consequent increase in ROS production. This high-level ROS generation promotes genetic instability in tumors, favors growth, chemotherapeutic escape, and evasion of senescence, all contributing to the increase in tumor malignancy. Therefore, the mitochondrion, where ROS is mainly produced, seems to play an important role in the tumorigenic phenotype.76 The mitochondrial DNA (mtDNA) encodes 13 of the electron transport chain proteins, and consequently, alterations in these coding genes lead to OXPHOS malfunction and inability to synthesize ATP and to reduce oxygen, which generates ROS. In the past decade, a wide spectrum of mutations in mtDNA has been identified in human cancers,77,78 including depletion of mtDNA. The lower content of copy numbers of mtDNA was associated with higher tumor malignancy among astrocytomas.79 Further analysis of mitochondrial genome and mitochondria biogenesis in different tumors may offer new insights for cancer therapy.
There are several ways to lower ROS levels, with glutathione being the major molecule involved, which eliminates ROS by accepting an electron and converting to its oxidized form glutathione disulfide. The enzyme glutathione reductase uses NADPH to reduce glutathione disulfide to glutathione. Low levels of glutathione/glutathione disulfide have been demonstrated in highly proliferative glioma samples.80 Thus, therapeutic targets to control ROS levels can also be exploited to selectively kill cancer cells.
Considering the current knowledge about cancer metabolism, it seems theoretically reasonable to target metabolic pathways for the control of cancer progression. However, no significant effect on tumor growth has been observed when the targets were applied as monotherapy.81,82 Nevertheless, these drugs have been proved to sensitize tumors to other chemotherapeutic agents (such as paclitaxel),83 presumably through reducing ATP levels. Then, new inhibitors of the glycolytic pathway have been used in combination with both chemotherapy and radiotherapy, in Phase II and III clinical trials against breast, ovarian, lung cancers,84-86 and GBM45 and the outcome of these trials may permit a better understanding of the metabolic impact on tumorigenesis. Specifically targeting the tumor cells and not normal proliferating cells continues to be an issue with this antimetabolic strategy for cancer control, as with most other chemotherapies. Targeting tumor-specific enzyme isoforms, suppressing the activity of an enzyme, or modifying the concentration of a substrate instead of completely abolishing it might provide a strategy to attack mainly tumor cells. Interestingly, the drugs developed to target metabolic diseases may also be applied in oncotherapy. Metformin, an oral hypoglycemic agent widely used to treat diabetes, is such an example.87,88 A number of retrospective clinical studies have found that metformin may offer a possible benefit in cancer prevention as well as in outcome when used with other cancer therapies.89 Similarly, an interesting premise is to bypass glycolysis through diet modification. The ketogenic diet with low carbohydrate and high fat, specifically medium-chain triglycerides, relies on the consumption of food that does not increase plasma glucose, but produces ketone bodies as a carbon source for energy production. Tumor cells depending on high glycolytic flux are not expected to survive on this alternative fuel source.90,91 Although this type of diet is not likely to be applied as a monotherapy, it may well prove to be useful to reinforce other antimetabolic therapies.
Recently, it was proposed that several “waves” of gene expression reprogramming occur during carcinogenesis, starting with oncogene-mediated alterations, followed by HIF-mediated expression, resulting in increased aerobic glycolysis and suppression of mitochondrial biogenesis. Then, on account of a high proliferation rate leading to nutrient shortage, there would be a third “wave” of adaptation with enhanced glutaminolysis. A retrograde signaling from revitalized mitochondria might constitute a fourth “wave”.92 Most probably, these proposed waves may occur simultaneously in a heterogeneous solid tumor, where hypoxic areas coexist with oxygenated areas, leading to a distinct pool of cells with different metabolic characteristics.
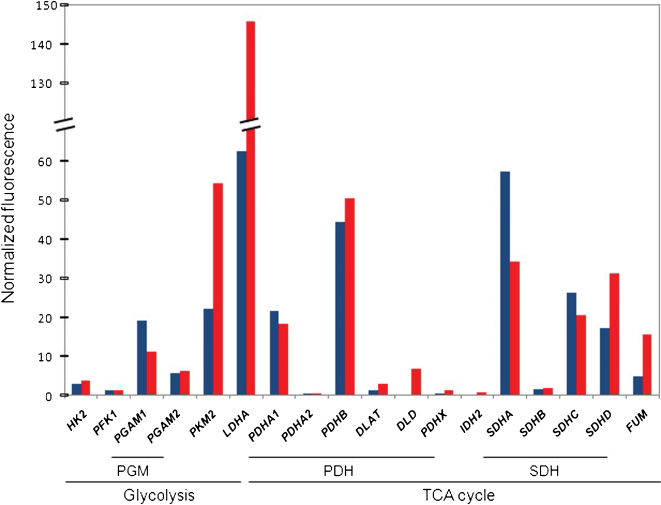
In fact, the results produced by our group in a series of primary GBM corroborate this heterogeneity.93,94 Concerning the metabolic profile of these GBM, the oligonucleotide microarray data have shown that all the key enzymes in the glycolytic pathway were not hyperexpressed and nor were the TCA cycle enzymes hypoexpressed. Figure 3 shows the mean normalized fluorescence of these key enzymes genes of three different samples of non-tumoral brain tissues and GBM. Concomitantly, depletion of the mitochondrial DNA with overexpression of the mitochondrial transcription factor A was observed, not following the theoretical model, confirming the complexity of mitochondrial biogenesis reprogramming.79 These preliminary results confirm the complexity of metabolism regulation of a tumor composed of pools of cells in different stages of metabolic reprogramming. Therefore, most probably, a better outcome for the cancer patient may be achieved by the better dynamic combination of targeted therapies.
 and three samples of GBM (red bars) were submitted to extraction of total RNA and microarray analysis, as described previously.94,146 LDHA and PKM2, both enzymes of glycolysis, are upregulated in GBM, whereas the variability in the expression profile of TCA cycle genes suggests that this cycle is uncoupled. The bars represent the median values of the three samples. Numbers represent the normalized fluorescence. Genes coding for glycolysis enzymes: HK2, hexokinase 2; PFK1, phosphofructokinase; PGAM1 and PGAM2, phosphoglycerate mutase 1 and 2, subunits of PGM dimer; PKM2, pyruvate kinase M2; LDHA, lactate dehydrogenase A. Genes coding for the TCA cycle enzymes: PDHA1 and PDHA2, pyruvate dehydrogenase alpha 1 and 2; PDHB, pyruvate dehydrogenase beta; DLAT, dihydrolipoamide-acetyltransferase; DLT, dihydrolipoamide dehydrogenase; PDHX, pyruvate dehydrogenase complex, component X, all six subunits of the enzymatic complex PDH; IDH2, isocitrate dehydrogenase 2; SDHA, SDHB, SDHC, and SDHD, SDH complex, subunits A, B, C, and D; and FUM, fumarate hydratase.")
Oligonucleotide microarray data of genes coding for key enzymes or subunits of enzymatic complexes of glycolysis and the TCA cycle. Three samples of non-tumoral brain tissues (blue bars) and three samples of GBM (red bars) were submitted to extraction of total RNA and microarray analysis, as described previously.94,146 LDHA and PKM2, both enzymes of glycolysis, are upregulated in GBM, whereas the variability in the expression profile of TCA cycle genes suggests that this cycle is uncoupled. The bars represent the median values of the three samples. Numbers represent the normalized fluorescence. Genes coding for glycolysis enzymes: HK2, hexokinase 2; PFK1, phosphofructokinase; PGAM1 and PGAM2, phosphoglycerate mutase 1 and 2, subunits of PGM dimer; PKM2, pyruvate kinase M2; LDHA, lactate dehydrogenase A. Genes coding for the TCA cycle enzymes: PDHA1 and PDHA2, pyruvate dehydrogenase alpha 1 and 2; PDHB, pyruvate dehydrogenase beta; DLAT, dihydrolipoamide-acetyltransferase; DLT, dihydrolipoamide dehydrogenase; PDHX, pyruvate dehydrogenase complex, component X, all six subunits of the enzymatic complex PDH; IDH2, isocitrate dehydrogenase 2; SDHA, SDHB, SDHC, and SDHD, SDH complex, subunits A, B, C, and D; and FUM, fumarate hydratase.
Moreover, these metabolic modifications should be contextualized in oncogene/tumor suppressor gene pathways already known. From the metabolic view, an oncogenic pathway will provide autonomous nutrient uptake and a proliferative metabolism program, in contrast to a suppressor pathway, which will prevent nutrient utilization for anabolic processes. Development of future therapies might also fit in this model, targeting self-limiting steps of nutrient production and metabolism.25,95
Oncogenes and tumor suppressor genes participating in the metabolic switchOncogenesThe phosphoinositide 3-kinase (PI3K) signaling pathway is one of the major pathways linked to the development of several tumors, including primary GBM.96,97 PI3K is recruited to the cellular membrane through the activation of epidermal growth factor receptor (EGFR), and it is phosphorylated from phosphatidylinositol-4,5-biphosphate to 3-phosphate, which activates target molecules such as protein kinase b (AKT) and mammalian target of rapamycin (mTOR), which induces cell proliferation and cell survival by blocking apoptosis. Several molecular participants in this pathway, such as EGFR, PTEN, and PIK3CA, present alterations in GBM.98
The PI3K/AKT pathway may regulate glucose metabolism by: (1) regulating glucose transporter expression through AKT, which directly stimulates transcription of the GLUT1;99 (2) enhancing glucose capture by HK2 and inducing aerobic glycolysis by promoting HK2 binding to voltage-dependent anion channels;100 and (3) stimulating PFK1 activity50 (Figure 2). In addition, activation of the PI3K pathway renders cells dependent on high levels of glucose flux.101 Small molecules inhibiting the PI3K pathway lead to decreased glucose uptake and correlate with tumor regression;102 response to therapy has been predicted by the ability to disrupt glucose metabolism measured by 18F-deoxyglucose positron emission tomography.103
AKT is activated when tumor cells need metabolic intermediates for rapid proliferation, and it promotes a glycolytic switch under normoxic conditions without affecting the rate of OXPHOS. Thus, cancer cells turn out to be dependent on aerobic glycolysis for their growth and survival under AKT-mediated metabolic influence; consequently, activated AKT tumor cells undergo rapid cell death when shifted to low-dose glucose conditions.104
HIF-1 is a pleiotropic hypoxia induced transcription factor with a heterodimeric structure composed of HIF-1α as the oxygen responsive subunit and HIF-1β as the constitutively expressed subunit.105 Hypoxia is a common microenvironment feature of solid tumors, leading to HIF-1 upregulation and consequently inducing a metabolic switch. HIF-1 target genes are involved in glucose and energy metabolism,106 including all glycolytic enzyme genes except phosphoglycerate mutase (PGM).107,108 Additionally, HIF-1 upregulates LDHA;109 PFK1, through activation of PFKFB3,110,111 switches the isoform PKM1 to PKM2112 and activates PDK1, which in turn inhibits PDH, reducing the TCA cycle.113 HIF also regulates the uptake of glucose through GLUT1 (Figure 2).114
MYC regulates a broad spectrum of genes involved in cell cycle control, metabolism, mitochondrial function, and regulation of apoptosis. However, the role of MYC is complex because its ectopic expression in cancer can concurrently drive aerobic glycolysis and/or OXPHOS according to the tumor cell microenvironment. MYC may trigger the expression of glycolytic genes or repress microRNAs miR-23a/b to increase GLS and stimulate OXPHOS through the activation of O2-dependent glutaminolysis (Figure 2).115 Then, MYC driven tumor cells are particularly sensitive to glutamine withdrawal,116 and genes involved in glutamine metabolism appear to be under both direct and indirect transcriptional control of the MYC protein.115,117 Glutamine depletion from MYC-transformed cells results in the rapid loss of TCA cycle intermediates and cell death.115 MYC also influences glycolysis by upregulating the majority of glycolytic genes, including HK2, PFK1, LDHA, PKM2, as well as GLUT1 (Figure 2).113 Deregulated expression of MYC collaborates with HIF-1 to confer the glycolytic (Warburg) phenotype of tumors. Additionally, MYC stimulates mitochondrial biogenesis in both normal and immortalized cells.118
Suppressor genesTumor suppressor pathways can also regulate cellular metabolism and may act to coordinate nutrient utilization with cell physiology.
TP53 is by far the most well known tumor suppressor gene. Inactivation of TP53 is observed in more than 50 different types of human cancer,119 and more than 27,000 mutations were enumerated in the International Agency for Research on Cancer – TP53 Mutation Database (http://www-p53.iarc.fr/). TP53 encodes a nuclear phosphoprotein of 53 kDa, responsible for genomic integrity and cell proliferation control. p53 protein facilitates DNA repair by blocking the cell cycle and induces apoptosis when the DNA damage is unrepairable.120–122TP53 mutation in germline cells determines Li–Fraumeni syndrome characterized by predisposition to multiple types of cancer, including breast tumor, sarcoma, and central nervous system tumors.123,124TP53 mutations are also frequent in secondary GBM.125–127 There are cumulative studies demonstrating a role for p53 in controlling metabolic genes, such as altering glucose utilization and promoting OXPHOS, but also contributing to the induction of apoptosis and maintaining mitochondrial health and activity by its localization inside the organelle. The most straightforward role of p53 in metabolism is the dampening of glycolysis, which is completely consistent with its function as a tumor suppressor. This limitation of glycolysis may occur in different ways: (1) downregulation of expression of several glucose transporters such as GLUT1 and GLUT4 by direct transcriptional repression128 and GLUT3 by the indirect reduction of expression through the inhibition of nuclear factor kappa-β kinase;129 (2) ubiquitination and inactivation of PGM;130 (3) lowering F2,6BP by p53-dependent expression of the TP53-induced glycolysis and apoptosis regulator (TIGAR).131,132 However, p53 also promotes glycolysis by increasing HK2 and PGM expression through p53-inducible promoters.133 Additionally, the increase in HK2 activity in concert with a decrease in PFK1 activity (resulting from TIGAR expression) would promote the use of the PPP.134,135 p53 has also been shown to play a role in promoting OXPHOS through the transcriptional activation of cytochrome c oxidase 2,136 subunit I of cytochrome c oxidase,137 and p52R2, a subunit of ribonucleotide reductase138 as well as by the post-transcriptional regulation of cytochrome oxidase 2.139 ATP is derived in much higher proportion through OXPHOS in cells expressing p53 than in cells lacking it.140 p53 also contributes to the maintenance of mitochondrial mass and mtDNA copy number.141,142
In addition to the complexity of tumor cell metabolic reprogramming in a context of impulses from oncogenes and inactivation of tumor suppressor genes, the cancer stem cell hypothesis should also be considered to gain a more complete understanding of tumorigenesis and for the efficacy of cancer therapy.
Targeting cancer stem cell metabolism as a future perspectiveThe cancer stem cell hypothesis proposes that cancer derives from a small fraction of cancer cells that constitute a self-sustaining cell reservoir, which are responsible for reseeding tumor cells after the first therapeutic approach. There are some preliminary data concerning the metabolism of the stem cells, such as increased expression of PFKFB3 in cycling cells and decreased expression in terminally differentiated neurons.143 It has been reported that GBM cancer stem cells share gene expression signatures with progenitor cells in the developing forebrain,144 and preferential overexpression of genes normally enriched in embryonic stem cells, NANOG, OCT4, SOX2, and MYC, were activated in poorly differentiated GBM.145 Moreover, a subpopulation of cells that carry the known progenitor cell marker CD133 has been identified in GBM, and when the expression profiles of CD133+ GBM cells were compared with CD133− cells, 16 genes, many of which had not previously been associated with astrocytomas, were found to be aberrantly expressed relative to corresponding non-neoplastic brain tissue controls.146 However, the expression profile of genes related to energy metabolism in cancer stem cells remains to be explored.
The development of novel metabolic based drugs holds much promise for the improvement of cancer therapy.