



To perform a molecular characterization of the C1q, C2 and C4 genes in patients with juvenile systemic lupus erythematosus.
METHODS:Patient 1 (P1) had undetectable C1q, patient 2 (P2) and patient 3 (P3) had decreased C2 and patient 4 (P4) had decreased C4 levels. All exons and non-coding regions of the C1q and C2 genes were sequenced. Mononuclear cells were cultured and stimulated with interferon gamma to evaluate C1q, C2 and C4 mRNA expression by quantitative real-time polymerase chain reaction.
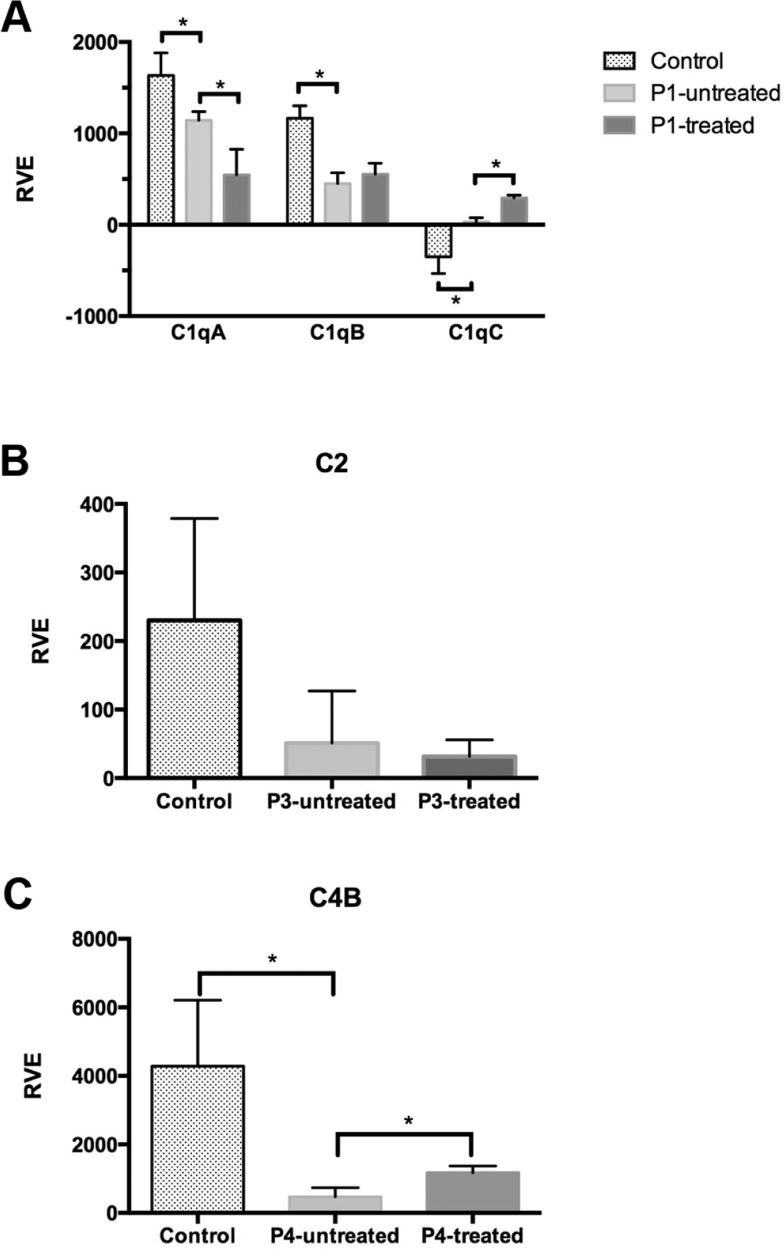
RESULTS:C1q sequencing revealed heterozygous silent mutations in the A (c.276 A>G Gly) and C (c.126 C>T Pro) chains, as well as a homozygous single-base change in the 3′ non-coding region of the B chain (c*78 A>G). C1qA mRNA expression without interferon was decreased compared with that of healthy controls (p<0.05) and was decreased after stimulation compared with that of non-treated cells. C1qB mRNA expression was decreased compared with that of controls and did not change with stimulation. C1qC mRNA expression was increased compared with that of controls and was even higher after stimulation. P2 and P3 had Type I C2 deficiency (heterozygous 28 bp deletion at exon 6). The C2 mRNA expression in P3 was 23 times lower compared with that of controls and did not change after stimulation. The C4B mRNA expression of P4 was decreased compared with that of controls and increased after stimulation.
CONCLUSIONS:Silent mutations and single-base changes in the 3′ non-coding regions may modify mRNA transcription and C1q production. Type I C2 deficiency should be evaluated in JSLE patients with decreased C2 serum levels. Further studies are needed to clarify the role of decreased C4B mRNA expression in JSLE pathogenesis.
Inherited deficiencies of the complement components C1q, C2 and C4 are frequently associated with the development of systemic lupus erythematosus (SLE) due to the impaired clearance of apoptotic debris, a decreased capacity for immune-complex handling, and/or their role in peripheral B-cell tolerance 1-9.
Homozygous C1q deficiency is the strongest genetic risk factor related to SLE 3,. Similarly, both Type I and II C2 deficiency have been associated with SLE 4,. In addition, homozygous C4A deficiency is approximately 10 times more frequent in patients with SLE 4,.
Few studies have evaluated the non-coding regions and mRNA expression of complement genes. Furthermore, complement deficiencies are rare among SLE patients but are common among patients with juvenile SLE (JSLE) 23.
We previously showed an underlying primary complement deficiency in seven JSLE patients 3. We had access to DNA samples and/or peripheral cells from four of these patients, and to better understand these inherited deficiencies, we conducted a molecular characterization of the C1q, C2 and C4 genes.
MATERIALS AND METHODSAmong 72 JSLE patients, 16 had primary immunodeficiency, and seven of the 16 were determined to have complement deficiency based on a combination of low or undetectable serum levels of C1q, or C1s, C1r, C2, C3 or C4 with normal levels of the other components in at least two samples and low activity/inactive disease (SLEDAI-2K score<4) 3. Therefore, we selected four patients: patient 1 (P1) had undetectable serum C1q, normal C3 and C4 levels, negative anti-C1q and available DNA and mononuclear cells; patient 2 (P2) had decreased IgA, C2 and C4 serum levels and stored DNA but no cells available for culture; patient 3 (P3) had decreased C2 and normal C3 and C4 levels; and patient 4 (P4) had repeated decreased C4 and normal C1q, C2 and C3 levels and peripheral cells available for culture (Tables 1 and 2). Three healthy controls matched by age and gender and with normal complement levels were also enrolled. Patients fulfilled the revised American College of Rheumatology classification criteria, and informed consent was obtained for all participants after the local ethics committee had approved the study 24,25.
-Complement component values from four juvenile systemic lupus erythematosus patients selected for molecular characterization from a cohort of seven patients previously determined to have primary complement deficiencies.
| Patient | C1q (33-209 mg/l) | C2 (14-25 mg/l) | C3 (0.5-1.8 g/l) | C4 (0.1-0.4 g/l) | C4 copy number |
|---|---|---|---|---|---|
| P1 | 0.0/0.0 (anti-C1q -) | 8.7 | 0.87 | 0.13 | Not done |
| P2 | 135.6 | 4.1/0.0 | 1.02 | 0.0/0.08 | 2 copies C4A 1 copy C4B |
| P3 | 4.6/12 (anti-C1q +) | 3.4/14 | 1.11 | 0.14 | Not done |
| P4 | 38.7 | 14.6 | 0.76 | 0.04/0.05 | 2 copies C4A 3 copies C4B |
*There was neither patient with C1r or C1s alterations, nor with persistent low C3 levels.
Clinical and laboratory findings from four juvenile systemic lupus erythematosus patients with primary complement deficiencies who were selected for molecular characterization 3.
| Patient/complement deficiency | Gender | Age at diagnosis (years) | Clinical and laboratory findings/Evolution |
|---|---|---|---|
| P1/C1q | female | 6 | Photosensitivity, membranous glomerulonephritis, positive ANA, anti-dsDNA, anti-Sm, anti-RNP and anti-La/lost for follow-up |
| P2/C2 | female | 13 | Diffuse proliferaitive glomerulonephritis, thrombocytopenia, positive ANA, anti-Ro, and anti-La. A brother with SLE and mother with Sjögren's syndrome/dead |
| P3/C2 | female | 6 | Serositis, arthritis, lymphopenia, photosensitivity, alopecia, oral ulcers, positive ANA and anti-dsDNA/follow-up |
| P4/C4 | female | 6 | Arthritis, serositis, seizures, hemolytic anemia, lymphopenia, positive ANA and anti-dsDNA/follow-up |
We previously determined the C4 gene copy number by quantitative real-time polymerase chain reaction (qRT-PCR), as reported 3,26 (Table 1).
All exons and 3′ and 5′ non-coding regions (UTR) of the C1q and C2 genes (3 exons each for the C1q A, B, and C chains and 18 exons for the C2 gene) were sequenced. Primers (Supplementary Table) were designed using the reference sequences deposited in GenBank (NG_007282.1 for C1qA, NG_007283.1 for C1qB, NG_007565.1 for C1qC and NG_011730.1 for C2) with the Primer-BLAST program. DNA was obtained using the QIAamp DNA Blood Midi kit (N°51104 Qiagen, Hilden, Germany), and purity and concentration were determined with a Nanovue® spectrophotometer (GE Healthcare, USA). Each PCR reaction was performed in a final volume of 25 µl containing 100 ng of DNA, 20 mM Tris-HCl (pH 8.4), 50 mM KCl, 2.0 mM MgSO4, 100 µM each of dATP, dCTP, dGTP and dTTP, 0.5 µM of each primer and 1.5 U of High-Fidelity Platinum Taq DNA Polymerase (Invitrogen, Carlsbad, CA, USA). The amplification conditions included 35 cycles of 95°C for 30 s, 60°C for 30 s and 72°C for 2 min, with an initial denaturing step of 95°C for 5 min and a final extension step of 72°C for 7 min. Products were purified using the GFX PCR DNA and Gel Band Purification Kit (GE Healthcare Life Science, UK). Amplicons and primers were sent to Centro de Estudos do Genoma Humano for sequencing. The BioEdit and ClustalW sequence alignment editors (http://www.mbio.ncsu.edu/bioedit/bioedit.html) were used to align the nucleotide sequences. The BLAST program (http://blast.ncbi.nlm.nih.gov/Blast.cgi) was used to search variant sequences obtained from the human genomic database. The Chromas software program was used for chromatogram visualization. Type I C2 deficiency was evaluated by PCR as previously described and confirmed by gene sequencing 3,27.
The quantitative analysis of C1q, C2 and C4 mRNA was performed using peripheral mononuclear cells obtained by centrifugation using Ficoll-Paque Plus (GE Healthcare, Piscataway, NJ, USA) and cultured in supplemented DMEM medium (10% fetal bovine serum, 10,000 U/ml penicillin and 100 g/ml streptomycin) at 37°C with 5% CO2 for 36 hours. Parallel cultures were stimulated with 100 U/µl of recombinant human interferon gamma (IFNγ; N°300-02 Preprotech, Rocky Hill, NJ, USA) as previously described 28. RNA was obtained from cultures using an RNeasy Midi Kit (N°74104, Qiagen, Hilden, Germany). RNA (500 ng) was reverse-transcribed to produce complementary DNA (cDNA) using the SuperScript III First-Strand Synthesis System for RT-PCR (N°18080-051, Invitrogen, Carlsbad, CA, USA). The qRT-PCR was performed using 2 µl of cDNA, 1 µM each of forward and reverse primers and 1X SYBR Green Mix QuantiFast PCR Kit (N°204054, Qiagen, Hilden, Germany) in a StepOnePlus Real-Time PCR System (Applied Biosystems, Forrest City, CA, USA). The cycling parameters included an initial 15 min hot start at 95°C, followed by 50 cycles of 15 s at 95°C and 30 s at 60°C. C4B primers were designed based on a GenBank reference sequence (NG_011638, Supplementary Table). Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) was used as an endogenous reference gene for qRT-PCR reaction normalization. Relative expression (RE) was determined by the standard curve method, and mean values are presented here as relative variation expression (RVE) obtained from RVE = target gene RE – GAPDH RE.
A t-test was used to compare the control and untreated samples and the patient treated and untreated samples. p<0.05 was considered significant.
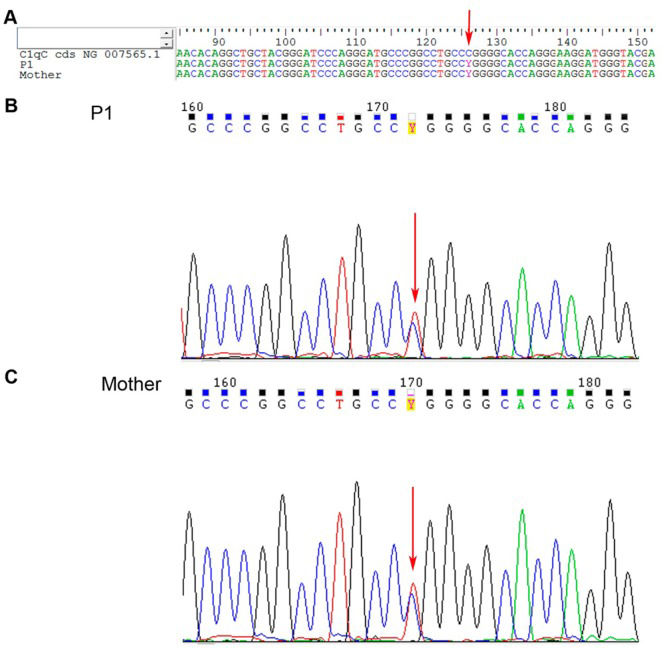
RESULTSC1q gene sequencing (P1) revealed a heterozygous silent mutation in exon 3 of the A chain (c.276 A>G Gly), a homozygous single-base change in the 3′ UTR of the B chain (c*78 A>G), and a heterozygous silent mutation in exon 2 of the C chain (c.126 C>T Pro) (Figures 1,2,3). The qRT-PCR analysis revealed that C1qA mRNA expression was decreased compared with that of healthy controls (p<0.05) and was also decreased after IFNγ stimulation (treated cells) compared with that of non-treated cells; C1qB mRNA expression was decreased compared with that of controls and did not change with stimulation, and C1qC mRNA expression was increased compared with that of controls and was even higher after IFNγ stimulation (Figure 5A).
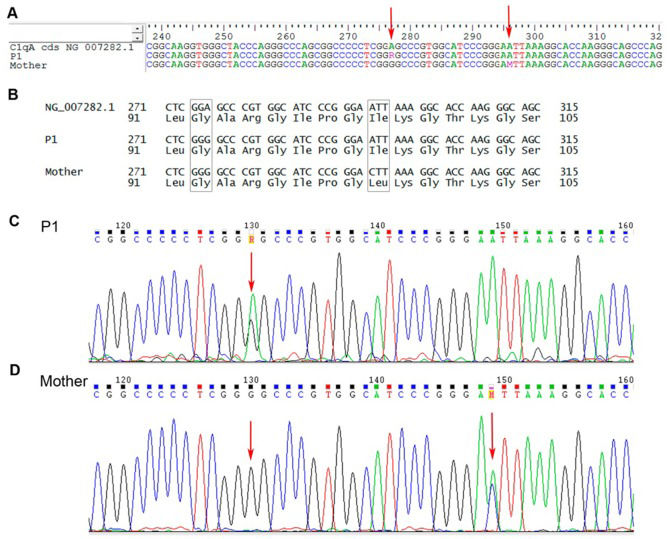
 G Gly; c.295 A>C Ile-Leu).' title='C1qA gene sequencing from juvenile systemic lupus erythematosus patient 1 (P1) and P1's mother. A – Nucleotide sequence alignment for P1 and P1's mother; B – Amino acid sequence for P1 and P1's mother; C and D – Nucleotide chromatograms of P1 and P1's mother, respectively. NG_007282.1 is the reference codified sequence for C1qA. The arrows indicate the variation sites (exon 3, c.276 A>G Gly; c.295 A>C Ile-Leu).'/>
G Gly; c.295 A>C Ile-Leu).' title='C1qA gene sequencing from juvenile systemic lupus erythematosus patient 1 (P1) and P1's mother. A – Nucleotide sequence alignment for P1 and P1's mother; B – Amino acid sequence for P1 and P1's mother; C and D – Nucleotide chromatograms of P1 and P1's mother, respectively. NG_007282.1 is the reference codified sequence for C1qA. The arrows indicate the variation sites (exon 3, c.276 A>G Gly; c.295 A>C Ile-Leu).'/>C1qA gene sequencing from juvenile systemic lupus erythematosus patient 1 (P1) and P1's mother. A – Nucleotide sequence alignment for P1 and P1's mother; B – Amino acid sequence for P1 and P1's mother; C and D – Nucleotide chromatograms of P1 and P1's mother, respectively. NG_007282.1 is the reference codified sequence for C1qA. The arrows indicate the variation sites (exon 3, c.276 A>G Gly; c.295 A>C Ile-Leu).
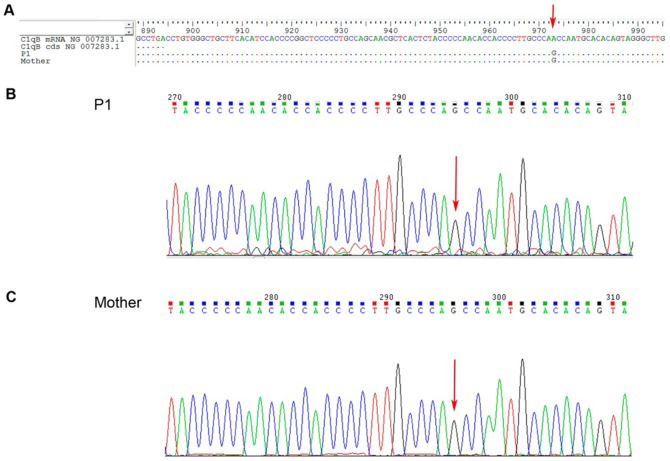
 G).' title='C1qB gene sequencing from juvenile systemic lupus erythematosus patient 1 (P1) and P1's mother. A – Nucleotide sequence alignment for P1 and P1's mother; B and C – Nucleotide chromatograms of P1 and P1's mother, respectively. NG_007283.1 is the reference codified or mRNA sequence for C1qB. The arrow indicates the variation site (3′ UTR c*78 A>G).'/>
G).' title='C1qB gene sequencing from juvenile systemic lupus erythematosus patient 1 (P1) and P1's mother. A – Nucleotide sequence alignment for P1 and P1's mother; B and C – Nucleotide chromatograms of P1 and P1's mother, respectively. NG_007283.1 is the reference codified or mRNA sequence for C1qB. The arrow indicates the variation site (3′ UTR c*78 A>G).'/>C1qB gene sequencing from juvenile systemic lupus erythematosus patient 1 (P1) and P1's mother. A – Nucleotide sequence alignment for P1 and P1's mother; B and C – Nucleotide chromatograms of P1 and P1's mother, respectively. NG_007283.1 is the reference codified or mRNA sequence for C1qB. The arrow indicates the variation site (3′ UTR c*78 A>G).
 T Pro).' title='C1qC gene sequencing from juvenile systemic lupus erythematosus patient 1 (P1) and P1's mother. A – Nucleotide sequence alignment for P1 and P1's mother; B and C – Nucleotide chromatograms of P1 and P1's mother, respectively. NG_007565.1 is the reference codified sequence for C1qC. The arrow indicates the variation site (exon 2, c.126 C>T Pro).'/>
T Pro).' title='C1qC gene sequencing from juvenile systemic lupus erythematosus patient 1 (P1) and P1's mother. A – Nucleotide sequence alignment for P1 and P1's mother; B and C – Nucleotide chromatograms of P1 and P1's mother, respectively. NG_007565.1 is the reference codified sequence for C1qC. The arrow indicates the variation site (exon 2, c.126 C>T Pro).'/>C1qC gene sequencing from juvenile systemic lupus erythematosus patient 1 (P1) and P1's mother. A – Nucleotide sequence alignment for P1 and P1's mother; B and C – Nucleotide chromatograms of P1 and P1's mother, respectively. NG_007565.1 is the reference codified sequence for C1qC. The arrow indicates the variation site (exon 2, c.126 C>T Pro).
. A – The relative expression of the C1q A, B and C chains in P1")
Relative expression of mRNA in cells from juvenile systemic lupus erythematosus patients and controls and after in vitro IFN-γ stimulation (treated). A – The relative expression of the C1q A, B and C chains in P1's cells; B – The relative expression of C2 in P3's cells; C – The relative expression of C4B in P4's cells. RVE – relative variation expression. *indicates a significant difference, p<0.05.
P1's healthy mother had an identical but homozygous silent mutation in the A chain, a homozygous single-base change in the B chain, a heterozygous silent mutation in the C chain, and an additional missense mutation in the A chain (c. 295 A>C Ile-Leu) (Figure 1). The peripheral cells of the mother were not available for mRNA expression.
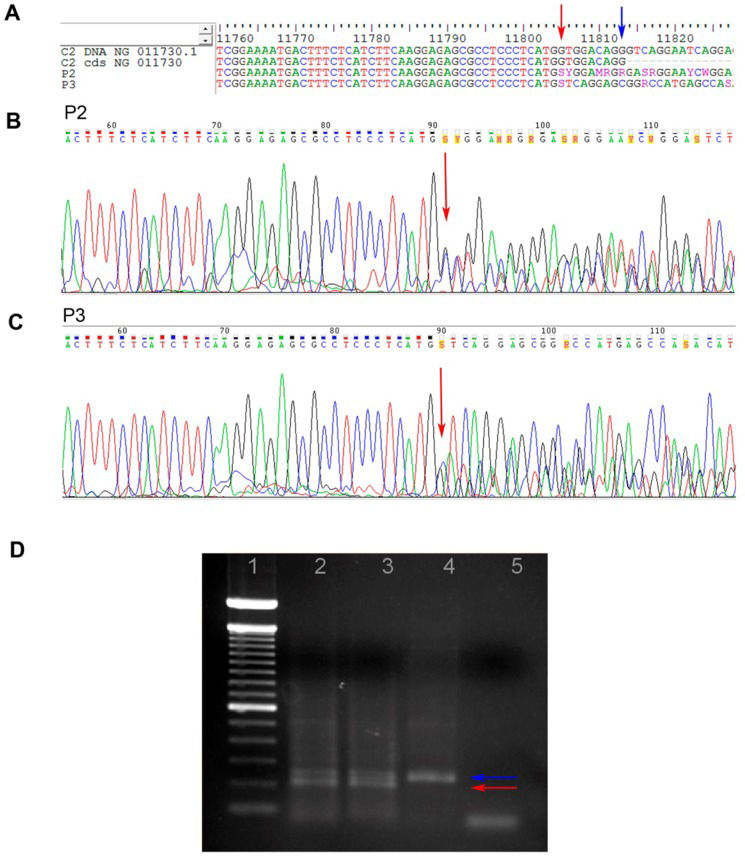
P2 and P3 had a heterozygous 28 bp deletion at exon 6, characterizing Type I C2 deficiency, as shown in Figure 4. The C2 mRNA expression (P3) was 23 times lower compared with that of controls and did not change after IFNγ stimulation (Figure 5B).
 and 3 (P3). A – Nucleotide sequence alignment for P2 and P3; B and C – Nucleotide chromatograms for P2 and P3, respectively. NG_11730.1 is the reference DNA sequence for C2, and NG_11730 is the codified sequence for C2 (final region of exon 6). The blue arrow indicates the last nucleotide of exon 6, and the red arrow indicates the initial deletion site; D – Agarose gel electrophoresis showing the heterozygous 28 bp deletion in exon 6 of the C2 gene. Lane 1: 100 bp DNA ladder; lane 2: P2; lane 3: P3; lane 4: positive control; lane 5: negative control. The blue and red arrows indicate the 174 bp and 146 bp fragments, respectively.")
Type I C2 deficiency in juvenile systemic lupus erythematosus patients 2 (P2) and 3 (P3). A – Nucleotide sequence alignment for P2 and P3; B and C – Nucleotide chromatograms for P2 and P3, respectively. NG_11730.1 is the reference DNA sequence for C2, and NG_11730 is the codified sequence for C2 (final region of exon 6). The blue arrow indicates the last nucleotide of exon 6, and the red arrow indicates the initial deletion site; D – Agarose gel electrophoresis showing the heterozygous 28 bp deletion in exon 6 of the C2 gene. Lane 1: 100 bp DNA ladder; lane 2: P2; lane 3: P3; lane 4: positive control; lane 5: negative control. The blue and red arrows indicate the 174 bp and 146 bp fragments, respectively.
The C4B mRNA expression in P4 was significantly decreased compared with that of controls and increased after IFNγ stimulation (Figure 5C). The C4A mRNA expression was not technically adequate, and it was therefore not considered for analysis.
DISCUSSIONFor the first time in JSLE patients, we showed heterozygous silent mutations, a homozygous single-base change in the 3′ UTR and decreased mRNA expression for the C1q gene; a heterozygous Type I deficiency and decreased mRNA expression for the C2 gene; and decreased mRNA expression for the C4B gene.
Although complement deficiencies were the first identified genetic risk factors for SLE, it remains unclear how these deficiencies should be assessed 2-4,7,8,27,.
Studies have described nonsense and missense mutations in the C1q gene, with a total of 12 causative mutations 3-5,10-12,23,. Single nucleotide polymorphisms and silent mutations have also been described 12,38. However, although there were no amino acid changes due to the identified base changes, the mRNA expression of the C1qA, B and C chains was altered. There is evidence that silent mutations can influence gene expression via alternative splicing by altering the exonic splicing enhancer or silencer sites 39. There is no evidence that heterozygous mutations of the C1q gene can lead to lupus if the other allele is normal. Of note, our patient presented photosensitivity, which is a characteristic associated with a single-nucleotide change in the C1qA gene 38. In addition, Rafiq reported allelic variance in the 3′ UTR of the C1qB chain associated with low C1q serum levels 36. The fact that the 3′ UTR single-base change in C1qB was homozygous implies that the mother and father each have one allele with the same sequence variants. P1's mother had normal C1q levels and did not present any symptoms of autoimmune disease; however, she harbored silent mutations with an additional missense mutation in the A chain. Because the C1q protein was present in P1's mother, it becomes questionable whether the observed variants are, by themselves, pathogenic 40,41. Peripheral cells from the patient's father and mother were not available for mRNA expression analysis, which could have addressed on the latter speculation.
An intriguing aspect of these results was the observation of mRNA expression from the C1q chains after IFNγ stimulation. IFNγ is able to induce and up-regulate complement production by monocytes 28,42,43. C1qA and B expression was decreased, and C1qC was increased, which suggests deregulated production that could influence the heterotrimeric assembly of protein. Nanjou et al. demonstrated that only one defect in any C1q chain can compromise the entire synthesis and secretion of the C1q protein 37. Because mRNA was still expressed, it is unlikely that the observed base changes could completely block protein expression, and alternative causes must be considered. Therefore, further work assessing mRNA stability and translation efficiency will be needed.
Type I C2 deficiency is due to a 9 bp deletion from the 3′ end of exon 6 and a 19 bp deletion from the 5′ region of the following intron, leading to a frame shift and a premature stop codon 2,13,15,16,29. More than half of individuals with primary C2 deficiency develop SLE or lupus-like disease 2,29. C2 gene sequencing was performed because most Type I deficiencies associated with SLE are homozygous and because Zhu et al. have reported a compound heterozygous Type I and II C2 deficiency in an African-American family 14-16,.
Although the Type I C2 deficiency was heterozygous, it resulted in a 23-fold reduction in mRNA expression. Lipsker et al. observed decreased but still detectable C2 levels in SLE patients with a heterozygous Type I C2 deficiency 43. Our findings raise both (1) the possibility that heterozygous C2 mutations alone or in combination with other factors could lead to SLE and (2) the question of why C2 mRNA expression was so low when the patient still had one functional C2 gene. Therefore, further JSLE patients with decreased C2 serum levels should be evaluated for Type I C2 deficiency to better understand the relationship of this condition with the disease.
The C4A and C4B genes each have 41 exons and differ from each other by only five nucleotides. The number of gene copies ranges from two to eight, and deficiencies in these two genes have been associated with SLE 18-20,44,. P4 had persistently low C4 serum levels, with two copies of C4A and three of C4B; the C4B mRNA expression in this individual was significantly decreased compared with controls and increased after IFNγ stimulation. Although this was an interesting finding, attributing the abnormal C4B mRNA expression to a gene mutation requires presuming that there were mutations in most or all of her three copies, which is possible but unlikely. Additionally, we would presume that there was a simultaneous problem with C4A transcription, which could not be evaluated. Thus, further work is needed to clarify the role of decreased C4B mRNA expression in JSLE pathogenesis.
This study was unprecedented because it involved the sequencing of all exons and non-coding regions of the C1q and C2 genes as well as the evaluation of mRNA expression. The limitations included the small number of patients, which created the need for experimental replicates, and the need for fresh mononuclear cells for mRNA expression analysis.
In conclusion, silent mutations and single-base changes in the 3′ non-coding region may modulate mRNA transcription and C1q production; Type I C2 deficiency should be evaluated in JSLE patients with decreased C2 serum levels; and further studies are needed to clarify the role of decreased C4B mRNA expression in JSLE pathogenesis.
AUTHOR CONTRIBUTIONSAll authors drafted, critically reviewed for intellectual content and approved the final version of the manuscript. Liphaus BL contributed to study conception and design, data analysis and interpretation and manuscript writing. Umetsu N contributed to data acquisition, analysis and interpretation. Jesus AA contributed to data acquisition, analysis and interpretation. Bando SY contributed to data analysis and interpretation and manuscript writing. Silva CA contributed to patient enrollment and data acquisition. Carneiro-Sampaio M contributed to study conception, data interpretation and manuscript intellectual content.
The authors gratefully acknowledge all of the patients for their kind participation. The authors also acknowledge Luis EC Andrade and Estela M Novak for their assistance with laboratory tests and mRNA expression data analysis.
Financial support: This study was supported by São Paulo Research Foundation (FAPESP) grant 2008/58238-4.
No potential conflict of interest was reported.