




Immunosuppressive therapy in pediatrics continues to evolve. Over the past decade, newer immunosuppressive agents have been introduced into adult and pediatric transplant patients with the goal of improving patient and allograft survival. Unfortunately, large-scale randomized clinical trials are not commonly performed in children. The purpose of this review is to discuss the newer immunosuppressive agents available for induction therapy, maintenance immunosuppression, and the treatment of rejection.
In the last two decades, significant developments in immunosuppressive therapies have provided transplant teams with more options for the care of transplant patients, with the promise of less toxicity and comparable or better efficacy. There are several potential target sites for immunosuppressive agents in transplantation, with examples demonstrating both established and new agents in Figures 1 and 2. The number of available immunosuppressive agents offers additional therapeutic strategies to provide better patient and allograft survival. The use of these newer agents in children has been limited in clinical studies, leading to off-label usage in pediatric centers based on small case studies. In addition, the long-term risks of newer immunosuppressive agents are not well documented and pose several challenges for clinicians.
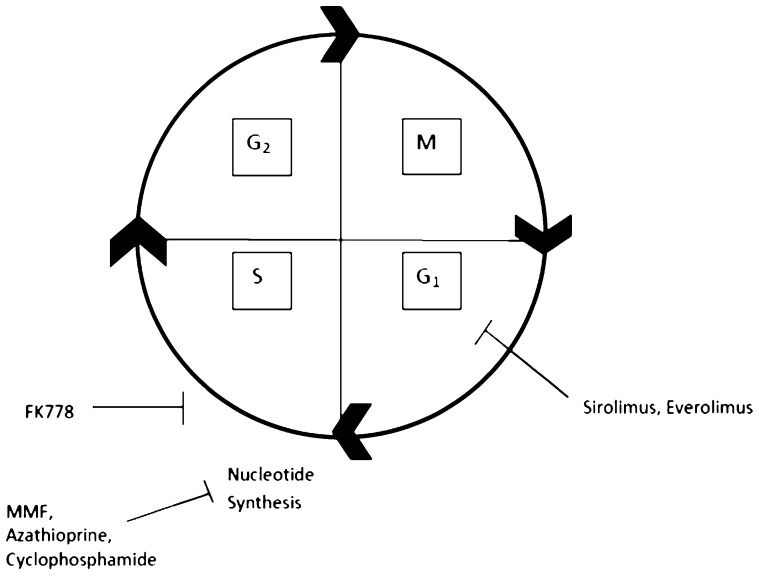
 includes prophase, metaphase, anaphase, and telophase. G1 is the first growth phase and includes growth and normal metabolic roles. The synthesis phase (S) is responsible for DNA replication. The second growth phase (G2) includes growth and preparation for mitosis. Sirolimus and everolimus inhibit the cell cycle at the G1 phase, while FK778 inhibits the cell cycle early in the S phase. MMF, azathioprine, and cyclophosphamide directly inhibit nucleotide synthesis in the S phase.")
Site of Action for Common Immunosuppression Agents in Cell Cycle. The mitotic phase (M) includes prophase, metaphase, anaphase, and telophase. G1 is the first growth phase and includes growth and normal metabolic roles. The synthesis phase (S) is responsible for DNA replication. The second growth phase (G2) includes growth and preparation for mitosis. Sirolimus and everolimus inhibit the cell cycle at the G1 phase, while FK778 inhibits the cell cycle early in the S phase. MMF, azathioprine, and cyclophosphamide directly inhibit nucleotide synthesis in the S phase.
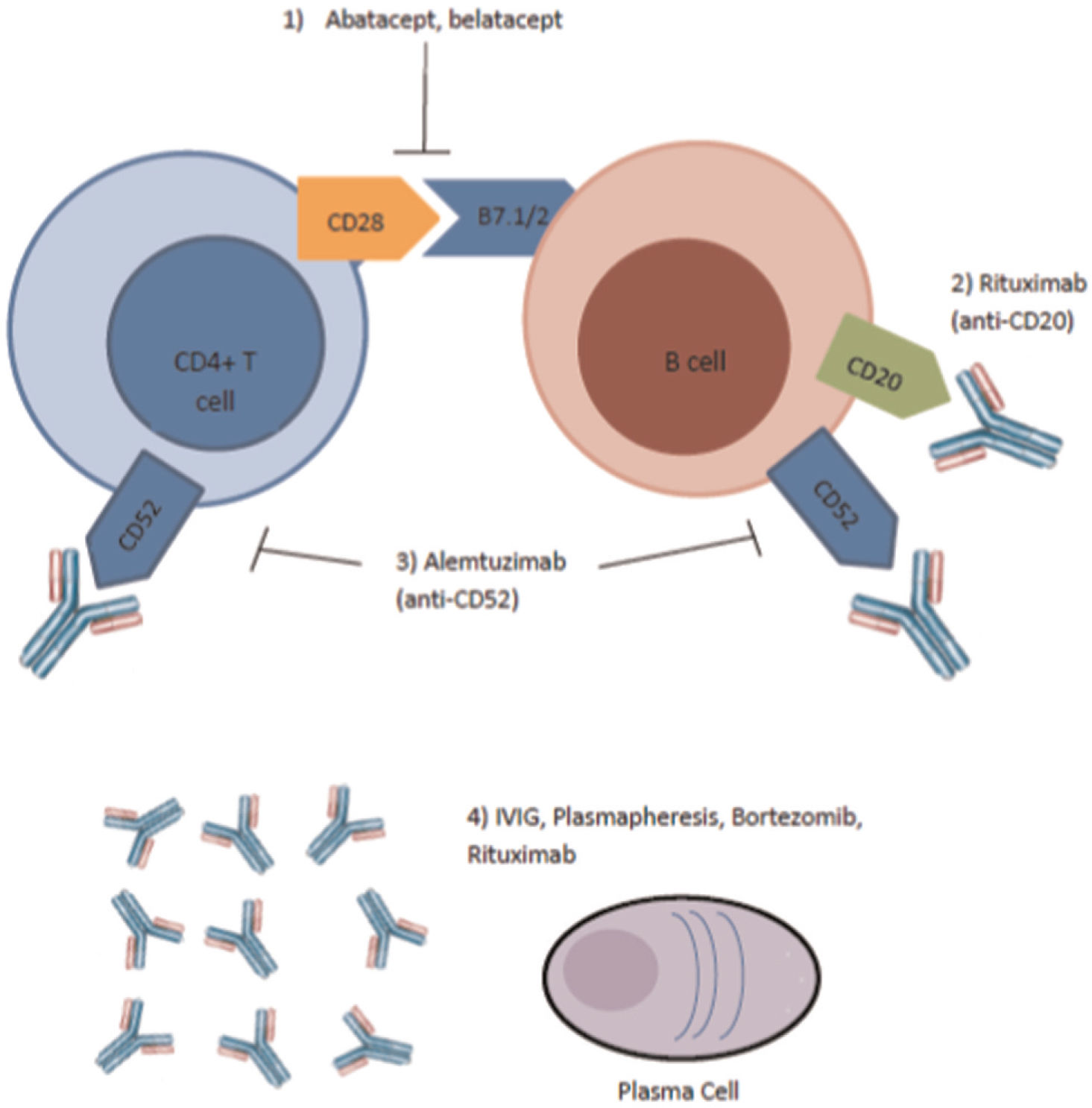
. Abatacept, belatacept, and alemtuzimab inhibit binding at the sites of T and B cells. IVIG, plasmapheresis, and rituximab affect the plasma cells.")
Site of Action of Newer Agents Used in Treatment of Antibody Mediated Rejection (39). Abatacept, belatacept, and alemtuzimab inhibit binding at the sites of T and B cells. IVIG, plasmapheresis, and rituximab affect the plasma cells.
Alemtuzumab, a monoclonal antibody targeting the CD52 antigen present on T and B lymphocytes, natural killer (NK) cells, and monocytes, was first used as an induction agent in kidney transplants by Calne et al. in 1998 (1,2). Although not approved by the United States Food and Drug Administration (FDA) for use in transplantation surgeries, it has gained popularity as an induction agent (1,3,4). An intravenous dose of 0.4-0.5 mg/kg, with a maximum dose of 30 mg, resulted in prolonged lymphocyte depletion (5,6). The effects of alemtuzumab include a profound depletion of total T cells and differential recovery of CD8+ T cells, with late and only partial recovery of the CD4+ subsets and relative sparing of CD4+ memory cells (7-9).
Serres and colleagues prospectively characterized the T cell subsets following lymphocyte depletion with alemtuzumab induction and the subsequent recovery patterns in children. Three months following alemtuzumab induction, both memory and naïve cells were depleted to a similar extent. Within the CD4+ memory subset, there was a slightly greater depletion of the central memory subset (TCM) compared with the effector memory subset (TEM), the latter of which is thought to be involved in acute cellular rejection (p = 0.002). At 24 months post-transplant, the recovery rate of total CD4+ T cells was slower than that of total CD8+ cells (p = 0.01), but there was no difference in the magnitude of recovery of naïve versus memory cells (p = 0.740). Of note, the recovery of TEM cells was nearly twice that of TCM cells, which is suggestive of an increased risk of late acute rejection (4). Interestingly, similar to the findings of Pearl et al., who studied an adult population, Serres found that children have an increased regulatory T-cell to TEM ratio in vivo, which may be tolerogenic (4,9). The percentage of children with circulating antibodies over the 2 years of the study was 23%, 50% of whom developed antibodies between 12 and 24 months (4).
Few studies exist on the use of alemtuzumab in children. Retrospective, small case studies have revealed equal efficacy between alemtuzimab induction and other induction agents, such as daclizumab and Thymoglobulin®, based on one-year actuarial patient and graft survival and renal function (5,10). Tan et al. retrospectively reviewed 42 consecutive living donor kidney transplantations performed over 4 years, in which the recipients received alemtuzumab prior to graft revascularization followed by tacrolimus monotherapy with spaced weaning. Of the 42 living donor transplants, 13 underwent transplant biopsies for a rising serum creatinine from baseline, and only 2 patients (4.8%) were diagnosed with acute cellular rejection (ACR). Every three months, the recipients were screened for anti-HLA antibodies and donor-specific antibodies were identified and characterized by Luminex assays. None of the patients in this study developed antibody-mediated rejection (AMR) (6).
There are certain settings in kidney transplantation where alemtuzumab may not necessarily be the ideal induction agent. In a small case series of complicated pediatric kidney transplant cases described by Bartosh and colleagues, alemtuzumab was associated with higher rates of rejection (11). In addition, the use of alemtuzumab may be problematic in kidney transplantations following a liver transplant in a hepatitis C virus-positive recipient or a recently transplanted patient who received heavy immunosuppression for the nonrenal organ, as this can be associated with higher rates of infection and a possible increased risk of post-transplant lymphoproliferative disorder (PTLD) (3).
SIROLIMUSThe introduction of cyclosporine in the 1980s dramatically transformed the field of transplantation. The success of calcineurin inhibitor (CNI)-based immunosuppression, however, is limited by nephrotoxicity. This limitation has led to an interest in CNI-free immunosuppressive regimens, but a solution remains elusive. Children with end-stage liver, intestinal, lung, and heart diseases are able to undergo organ transplantation, but the long-term consequences following non-renal solid organ transplantation include chronic kidney disease, end-stage renal disease, and the need for future kidney transplantation (12). The withdrawal of CNIs after a period of time after transplantation and conversion to CNI-free regimens have been attempted with mixed results (13-15). In recent years, there have been promising new agents that have become available for clinical use.
Sirolimus is a macrolide compound derived from Actinomyces hygroscopicus. It is structurally similar to tacrolimus and was introduced for clinical use in organ transplantation in 1999 (16). Sirolimus binds to the mammalian target of rapamycin (mTOR) in T cells, suppressing T-cell proliferation by inhibiting the progression from the G1 to S phase of the cell cycle. Sirolimus allows T cell activation but prevents the cells from proliferating in response to IL-2, which inhibits growth factor-induced proliferation of lymphocytes and cells of mesenchymal origin. Sirolimus has anti-proliferative effects on fibroblasts, endothelial cells, and smooth muscle cells that make it a promising agent for the prevention of chronic allograft nephropathy and the progression of kidney disease in non-renal solid organ transplantation (15,17-23).
Sirolimus is a hydrophobic agent that has rapid bioavailability. Variable effects can occur depending on food consumption, with oral suspension bioavailability decreasing with high-fat meals but increasing if tablets are administered. It is metabolized by the cytochrome P450 3A isoenzyme in the intestinal wall and liver. The half-life of sirolimus in adult renal transplant patients is 62±16 hours, but small trials have demonstrated more rapid clearance in younger children (16,24).
The most common side effects of sirolimus are dose-related and include hyperlipidemia and a variety of cytopenias. Interstitial lung disease and mouth ulcers have also been reported in adult and pediatric patients treated with sirolimus. Although sirolimus is not generally thought to be nephrotoxic, it can be associated with proteinuria, which can lead to nephrotic syndrome and subsequent kidney injury (25).
The use of sirolimus in children has not been well described, with most of the available clinical information originating from single-center case studies. Many groups are attempting CNI minimization and avoidance protocols with mTOR inhibitors, such as everolimus or sirolimus, to prevent post-transplant CKD. Chinnock and colleagues minimized CNI exposure by adding sirolimus to the immunosuppression protocol when pediatric heart transplant recipients developed renal insufficiency (GFR<60 mL/min/1.73 m2) and found that renal function improved or stabilized following CNI minimization (20). Hynes and colleagues examined the use of sirolimus-based calcineurin inhibitor-free immunosuppression in renal transplantation. They found that sirolimus-based immunosuppression could be successfully used in low-risk patients and first-time transplant recipients without histological evidence of acute rejection on a three-month surveillance biopsy. After five years, they found that apthous ulcers and BK virus viremia were the most prevalent adverse events (26). The role of sirolimus in CNI minimization is also being explored in other solid organ transplantations. Gibelli and colleagues found that conversion from CNI to sirolimus was safe, with no increased rates of rejection following pediatric liver transplantation (21).
EVEROLIMUSEverolimus is a macrocyclic lactone that was first isolated from Streptomyces hygroscoicus. It is a derivative of sirolimus and shares its mechanism of action. The advantages of everolimus are that it is more hydrophilic, demonstrating a shorter half-life of approximately 30 hours, and has greater bioavailability compared with sirolimus. Everolimus is metabolized by the cytochrome 450 3A isoenzymes and, similar to sirolimus, is also affected by dietary changes (16).
Based on experimental animal models and experience in cardiac transplantation, everolimus also appears to have antiproliferative effects on vascular smooth muscle cells and fibroblasts (16,27). The antiproliferative action of everolimus in the arterial wall has been shown to inhibit atherogenic and intimal wall remodeling in pre-clinical studies. It is also involved in a critical part of the cell cycle that has significant antineoplastic potential as a promoter of apoptosis and inhibitor of angiogenesis (28).
Ettenger and colleagues used a CNI minimization protocol comprising everolimus, cyclosporine, and corticosteroids following kidney transplantation. Of the 19 pediatric patients enrolled, 15 were followed for 3 years. The patient survival rate was 100%, and the allograft survival rate was 93%. Three patients had infectious complications, 4 patients developed rejection, 4 patients required treatment with statins for hypercholesterolemia, and the average serum creatinine was 1.1 mg/dL after 3 years (29). Another prospective study examined 20 children who received basiliximab, cyclosporine A, and prednisolone for immunosuppression. Two weeks following kidney transplantation, everolimus was initiated and cyclosporine A was reduced by 50%. Pape and colleagues found that the mean GFR at 1 and 3 years was 71 and 61 mL/min/1.73 m2, respectively. There were no cases of post-transplant lymphoproliferative disorder, acute rejection, or polyoma nephropathy, and 85% of the patients remained on the original immunosuppressive regimen (30).
Although only limited pediatric studies have been performed to evaluate the efficacy of everolimus in liver and heart transplantation, the early adult data are promising. A study in pediatric heart transplant recipients showed that conversion to CNI-free immunosuppression with everolimus and mycophenolate when the GFR fell below 75 mL/min/1.73 m2 resulted in improved renal function within 6 months of CNI withdrawal in pediatric heart transplant recipients (31). The possible benefit of the anti-fibrotic effects of everolimus, as well as its lack of nephrotoxicity, make it a promising immunosuppressive agent in pediatric transplantation (28).
BELATACEPTBelatacept is a biological agent that is a selective co-stimulation blocker of T cells. It is a fully human fusion protein of the CTLA4 extracellular domain with fragments of the Tc domain of human IgG1, and it has a binding affinity to CD86/CD80 on the antigen-presenting cell, thus resulting in the blockade of T-cell activation (32,33). The activation of T cells involves the presentation of a peptide by an antigen-presenting cell to the T cell receptor, followed by costimulatory associations between the ligands on the antigen-presenting cell and T cell receptors (33). In addition, belatacept indirectly inhibits antigen-specific antibody (IgG, IgM, and IgA) production by B lymphocytes, resulting in lower mean immunoglobulin concentrations compared with that resulting from cyclosporine-based therapy (32). Belatacept requires intravenous infusion, and the total infusion dose is based on the body weight of the patient. A dose of 10 mg/kg is administered on the day of transplantation, day 5, and at 2, 4, 8, and 12 weeks, followed by 5 mg/kg at the end of week 16 and every 4 weeks thereafter for maintenance (32,34).
The BENEFIT trial has resulted in the publication of data on adult kidney transplant recipients, including three year outcome data. BENEFIT was a randomized, three-year, phase III study in adults receiving either a standard deceased or living donor kidney transplant. The patients were randomized to a more intense regimen (MI), a less intense regimen (LI), or cyclosporine. Of the 666 patients who completed at least 3 years of therapy, 92% of the MI, 92% of the LI, and 89% of the cyclosporine patients survived with a functioning allograft. At 3 years, the average calculated GFR was 21 mL/min/1.73 m2 higher in the belatacept group compared with the cyclosporine group. This difference in GFR between the belatacept and cyclosporine groups was enough to classify the mean GFR as stage II (65 mL/min/1.73 m2) verses stage III (44 mL/min/1.73 m2) chronic kidney disease. Overall, stage II or better chronic kidney disease represented 69% of the MI, 71% of the LI, and only 29% of the cyclosporine-based treatment groups at 3 years. In addition to improved renal function, donor-specific antibodies occurred in 6% of MI, 5% of LI, and 11% of the cyclosporine treatment arms. The cumulative rates of acute rejection were 24% in the MI, 17% in the LI, and 10% in the cyclosporine group at year 3. Despite an early increased occurrence of acute rejection and post-transplant proliferative disorder, especially in EBV seronegative patients, belatacept-treated patients maintained a comparable rate of patient and graft survival with cyclosporine but had better renal function (35).
To date, clinical trials using belatacept have been conducted exclusively in adult patients, and no clinical trials in children have been listed through the National Institute of Health (36). Abatacept is a first-generation biological agent with a similar mechanism of action to belatacept, but it is used in patients with autoimmune disorders and not transplant patients. The pharmacokinetics of either abatacept or belatacept have not been studied specifically in children or adolescents. The efficacy of abatacept in the treatment of juvenile rheumatoid arthritis was studied by the Pediatric Rheumatology International Trials Organization and the Pediatric Rheumatology Collaborative Study Group in a population with ages ranging from 6-17 years. The trial was a randomized, double-blind, placebo-controlled withdrawal study that was performed at 45 centers in Europe and the United States between 2004 and 2006 and enrolled 190 patients. Several adverse events were documented, including a total of 95 infections in the open-label, lead-in, and controlled withdrawal phases and 1 serious infection secondary to varicella zoster. In the open-label, long-term extension phase spanning an average of 35 months, there were 6 serious infections, including dengue fever, erysipelas, gastroenteritis, herpes zoster, bacterial meningitis, and pyelonephritis. EBV or CMV disease was not mentioned (37). In 2009, the United States Food and Drug Administration (FDA) examined the 5-year post-marketing data of 90 patients treated with abatacept. Six serious adverse events were reported, four of which occurred in the United States. One case of multiple sclerosis (MS) and lymphoma occurred one month after starting abatacept, and the other serious events included skin infection, dyspnea, purpura, and transaminitis. The child who developed MS also developed temporal lobe epilepsy 19 months after starting abatacept for the treatment of JIA. He was also on methotrexate and ondansetron, which have been associated with the development of epilepsy. The child who developed lymphoma initially developed lymphomatoid papulosis 1 month after starting abatacept for vasculitis and had been on immunosuppressive therapy for 10 years (38).
Although belatacept has not been used in pediatric transplantation, it may have a role in the future. The method of administration may positively affect rates of medication administration non-compliance, which is a particular problem in adolescent and young adult transplant management. The higher rates of PTLD were concentrated in patients who were seronegative for Epstein-Bar virus (EBV) and those who were in the MI arm (35). This may be problematic for the extension of belatacept in pediatric patients, as many of these patients are seronegative for EBV at the time of transplantation.
BORTEZOMIBHistorically, T-cell-mediated mechanisms have been considered to be the main cause of allograft rejection. However, the role of humoral responses, particularly those mediated by alloantibodies, has increasingly being implicated in allograft rejection (39). As antibody-mediated rejection (AMR) has gained recognition, so has the realization that traditional therapies for AMR, including IVIG, plasmapheresis, rituximab, and antilymphocyte preparations, are inconsistent in efficacy (40).
Bortezomib is a proteasome inhibitor. The 26S proteasome is the site of degradation of nearly all intracellular proteins. Damaged and misfolded proteins, as well as proteins targeted for degradation, are all degraded in the proteasome. Proteasome inhibition induces endoplasmic reticulum (ER) stress, unfolded protein response (UPR), and terminal UPR. The UPR involves the inhibition of translation and, therefore, immunoglobulin production, and it also increases the cellular capacity to repair misfolded or unfolded proteins. Proteasome inhibition also induces programmed cell death by cell cycle arrest in cells undergoing mitosis at specific checkpoints (40). In vivo studies have demonstrated that proteasome inhibitors can result in the deletion of non-transformed plasma cells, which manufacture alloantibodies (40).
Bortezomib is approved by the FDA for the treatment of multiple myeloma (40). A variety of protocols exist for treatment of AMR with boretzomib, and examples are listed in Table1. In transplant patients, it was initially studied for the treatment of refractory AMR. Bortezomib has been demonstrated to reduce donor-specific antibodies in both heart and kidney transplant patients (40-42). Everly and colleagues used bortezomib to treat patients with refractory renal allograft rejection and found that bortezomib therapy was associated with improved renal function and renal allograft histology, as well as reductions in DSA levels. To remove circulating antibodies, plasmapheresis is necessary in addition to bortezomib administration (40,43). Utilizing rituximab at the start of treatment was found to reduce plasma cell antibody production prior to depletion with bortezomib (40). Morrow and colleagues treated four pediatric heart transplant patients with refractory AMR, who had failed plasmapheresis, intravenous immunoglobulin, and rituximab, with intravenous bortezomib, rituximab, and plasmapheresis. The patients had improved systolic function, and conversion to C4d-negative biopsies was observed in 75% of patients. Additionally, 25% of patients became IgG-negative and had a prompt and precipitous reduction in DSAs (44). Although studies have demonstrated the effectiveness of bortezomib, there are several case reports and small case studies that revealed bortezomib to be ineffective in prolonging graft survival, despite decreasing the DSA load, in prolonged or refractory AMR (45,46).
Bortezomib Protocols for the Treatment of Antibody-Mediated Rejection.
| Authors | Day | Bortezomib AMR Protocol |
|---|---|---|
| Morrow et al.(heart) (44) | 11,4,7,1114-1618Follow-up after 30 days | Rituximab 375 mg/m2 IVPlasmapheresis, Methylprednisolone (5 mg/kg), bortezomib (1.3 mg/m2 IV)PlasmapheresisPRADSA, T/B cell subsets |
| Cincinnati(kidney, pancreas, intestines, heart) (40) | 11,4, 8, 11, and 1416-19 or16-21 | Rituximab (375 mg/m2)Plasmapheresis (1.5 plasma volume), bortezomib (1.3 mg/m2), DSA. Biopsy at 8 and 14Plasmapheresis (1.5x plasma volume) daily, DSA day 19Plasmapheresis (1.5x plasma volume) every other day, DSA day 21 |
The University of Cincinnati developed a collaborative study group of transplant centers, the START collaboration, and has studied the use of bortezomib in over 91 solid organ transplants with 107 episodes of AMR. The experience included both adult and pediatric kidney, pancreas, and intestinal, as well as pediatric and adult heart, transplant recipients. Bortezomib provided effective treatment in all types of solid organ transplants studied (40).
ECULIZUMABAtypical hemolytic uremic syndrome (aHUS) is a disease of the microvasculature characterized by hemolytic anemia, thrombocytopenia, and acute kidney injury, with more than 50% of patients progressing to end-stage renal disease (ESRD) (47,48). The diagnosis of aHUS is made based on a lack of associated disease, criteria for Shigatoxin-HUS, and criteria for thrombotic thrombocytopenic purpura (TTP), with serum ADAMTS 13 activity >10%. Onset can vary from infancy to adulthood. Additionally, 2-10% die, 1/3 progress to ESRD after the first episode, and 50% of the patients have relapses. There is a high risk of post-transplant recurrence, except in the setting of membrane cofactor protein disease, in which case most patients have preserved renal function. The most frequent mutations are located in complement factor H (CFH), and the disease recurs in nearly 80% of patients transplanted with CFH mutations (47,49).
The known pathogenesis of aHUS is primarily related to the dysregulation of the complement cascade. In approximately 50% of affected patients, mutations in genes encoding complement proteins result in impaired regulation of the complement alternative pathway, leading to inappropriate complement activation in platelets and endothelial cells, primarily within the kidney (50). Until recently, renal transplantation alone has not been an effective treatment option for aHUS patients because of high morbidity and mortality rates primarily associated with disease recurrence and premature liver failure secondary to uncontrolled complement activation (48,50). Several combined liver-kidney transplants have been performed with the goal to have the transplanted liver supply normal complement regulatory proteins in patients with aHUS. However, this approach does not address the problem in patients with CFH autoantibodies (48,51-54).
Kidney transplantation markedly activates the alternative pathway of the complement system, resulting in a poor outcome in aHUS patients with mutations in FH (50). The identification of anticomplement therapy has led to a safer approach than kidney transplantation alone. Eculizumab is a complete, humanized C5 monoclonal antibody that inhibits complement factor 5a, blocking terminal complement activation and the formation of membrane attack complexes (48-50). The prophylactic use of eculizumab and plasmapheresis in kidney transplant recipients with aHUS has demonstrated favorable outcomes in several case reports involving genetic defects, including CFH, C3, complement factor H-related proteins (CFHR1 and CFH hybrids) (48,50,55-57).
There is also growing interest in using eculizumab to prevent complement-mediated injury in kidney transplantation, ranging from use in the positive cross-match donor to prevention of antibody-mediated rejection in sensitized recipients to the prevention of delayed graft function in deceased donors. Currently, several adult studies are actively recruiting participants (36). Eculizumab was used as a rescue treatment for severe AMR or prevention of AMR. Early results from the Mayo Clinic showed that eculizumab prevents the development of acute AMR in cross-match-positive patients. Eculizumab was added to the desensitization protocol with mixed results. One of 16 patients developed AMR, and 6 patients developed chronic AMR by 3.8 months post-transplantation (58-60).
FK778FK778 (manitimus) is a synthetic malononitrilamide derivative of the active leflunomide metabolite, teriflunomide, which has been demonstrated to have both immunosuppressive and anti-proliferative activities. It inhibits both T-cell and B-cell functions by blocking de novo pyrimidine synthesis, inhibiting tyrosine kinase activity, and suppressing IgG and IgM antibody production, as shown both in vitro and in vivo (16,61). In addition, FK778 prevents vascular remodeling after intimal injury and blocks the replication of the herpes virus family in vitro (16). Animal studies examining the role of FK778 in liver regeneration and acute rejection have been carried out in rats that underwent segmental liver transplantation. FK778 was found to be antihepatotrophic and antiproliferative in rat liver regeneration (62). FK778 was also found to prevent acute allograft rejection and reverse ongoing rejection in rats undergoing liver, heart, or kidney transplantation. Combination therapy with tacrolimus was also found to be more beneficial than FK778 monotherapy (63,64).
A phase II, multi-center, randomized, double-blind study compared FK778 with tacroilmus and steroids verses MMF with tacrolimus and steroids in renal transplantation patients and found that at low levels, FK778 and MMF had similar efficacy (61). Unfortunately, as there were not sufficiently good results to justify phase III studies of FK778, this agent is not being further developed.
JAK3 INHIBITORSThe Janus kinase family is a group of four cytoplasmic, non-receptor tyrosine kinases. JAK3 is highly expressed in lymphoid cells and is activated only by cytokines that bind to gamma chain-containing receptors. It is a potential immunosuppressive target because JAK3 activation leads to the dimerization of the STAT 5 transcription factor, which is specific for IL-2 cytokines. JAK3 controls various cytokine-regulated signal transduction pathways, including lymphocyte proliferation, differentiation, and apoptosis (65). Concerns regarding JAK3 inhibition exist because the loss of NK cells may lead to impaired innate immunity and memory. Additionally, as it is very similar to JAK2, which mediates signaling of hematopoiesis, its inhibition could potentially lead to anemia, leukopenia, and thrombocytopenia (16).
CP-690550, tofacitinib, is an oral JAK3 inhibitor with the potential to prevent transplant rejection and treat autoimmune diseases. Phase I and II clinical trials have demonstrated that tofacitinib is efficacious and safe in preventing organ transplant rejection (66). In a phase 2b trial, tofacitinib was equivalent to cyclosporine in preventing acute rejection and was associated with improved renal function and less chronic allograft disease. At the dose studied, tofacitinib caused side effects, and adverse events occurred in over 10% of the treatment group. Adverse events included serious infection and CMV disease, post-transplant proliferative disorder, anemia, and neutropenia. One anticipated challenge will be the monitoring of drug levels, and a decision has not yet been made to start phase III trials.
FTY 720FTY720, fingolimod, is derived from a fungal sphingosine-like metabolite. It is a sphingosine 1-phosphate receptor modulator that is able to entrap lymphocytes in secondary lymphoid organs, thus reducing their availability for cell-mediated immune responses in allografts (16,67). Fingolimod use has been associated with bradycardia, macular edema, and increased airway resistance. Additionally, when combined with tacrolimus, it did not demonstrate a significant therapeutic advantage over MMF in preventing acute rejection in kidney transplant recipients (16,68). Although it has not moved forward in the treatment of transplant recipients, it may have a role in treating patients with autoimmune diseases.
ALEFACEPTAlefacept, LFA-3-Ig, is a dimeric fusion protein consisting of the CD2-binding portion of the human lymphocyte function-associated antigen-3 (LFA-3) linked to the Fc portion of human IgG1. It is thought to neutralize the effect of CD2-expressing T-cells by complement-mediated lysis, interrupt the CD2 interaction with LFA-3, limit helper T-cell adhesion to antigen-presenting cells, and disrupt effector T-cell receptor engagement with antigens and major histocompatibility complex molecules (69). A pilot study assessing the safety and efficacy of alefacept in de novo kidney transplant recipients was terminated because of increased risks that were greater than expected based on the standard of care. However, alefacept is currently undergoing clinical trials for the prevention of graft versus host disease (16,36).
ASKP1240ASKP1240 is a novel, human anti-CD40 monoclonal antibody that is currently in pre-clinical trials as a new immunosuppressive agent that may promote tolerance induction in organ transplantation. ASKP1240 has been demonstrated to prolong renal allograft survival in non-human primates (NHPs), although the allografts eventually underwent chronic allograft nephropathy. Oura and colleagues completed a two-week ASKP1240 induction treatment in NHPs and found that although it prolonged liver allograft survival, the function of the graft deteriorated due to chronic rejection. Following a six-month ASKP1240 maintenance monotherapy protocol, cellular and humoral alloimmune responses were suppressed, rejection was prevented, and no serious side effects were observed in hepatic allograft recipients (70).
A clinical trial assessing the bioavailability of ASKP1240 in healthy subjects after intravenous and subcutaneous administration was recently completed in September 2012 and demonstrated that ASKP1240 was well tolerated in the range of 50 to 500 mg, with no evidence of cytokine release syndrome or thromboembolic events. There was no difference in the incidence of infection based on dose, but B cell CD40 receptor occupancy trended to be prolonged as the dose increased (71). Phase II trials are currently in development (36).
CMX001CMX001 is a novel, broad-spectrum lipid antiviral conjugate that produces high levels of the active antiviral agent cidofovir diphosphate and is active against multiple double-stranded DNA viruses, such as JC and BK viruses (72). It is a lipid conjugate of the acyclic nucleotide phosphonate cidofovir. The utility of cidofovir is limited by the high incidence of acute kidney toxicity and intravenous administration. Compared to cidofovir, CMX001 has greater potency in vitro against all double stranded DNA viruses that cause human disease, a higher genetic barrier to resistance, no evidence of nephrotoxicity in humans or animals, and is available as an oral agent (73).
Painter and colleagues conducted a randomized, double-blind, placebo-controlled, parallel-group, dose-escalation study in healthy volunteers to examine the pharmacokinetics and safety of CMX001. They found that CMX001 is orally bioavailable and well tolerated in healthy volunteers at doses of up to 2 mg/kg, which corresponds to approximately 140 mg in a typical adult (72). BKV is associated with nephropathy, as well as hemorrhagic cystistis (74). Treating polyomavirus BK (BKV) is of notable importance in kidney transplant recipients, as current agents, with the exception of immunosuppressive reduction, which places transplant recipients at increased risk for rejection, have not been consistently effective in reducing BK viral loads. Comparisons of CMX001 and cidofovir have demonstrated that CMX001 had more rapid and enduring effects on BKV DNA than cidofovir at lower levels and had fewer side effects on relevant host cells in vitro (74). No human studies in transplant recipients with BK nephropathy or viremia have been published to date, although an anti-viral agent against BKV without associated nephrotoxicity is promising and could positively impact the management of BKV.
CONCLUSIONDespite the advances in immunosuppressive therapy, the optimal immunosuppressive regimen remains elusive. Over the years, research in immunosuppressive therapy has intensified, and several promising agents targeting different sites of the immune system are under development. Important goals in finding new agents include reducing nephrotoxicity; preserving both short- and long-term graft function; and reducing the risk of opportunistic infections, PTLD, malignancy, and other side effects. These goals are particularly critical in pediatric transplant recipients due to their life expectancy and the need to prevent additional chronic medical conditions.
AUTHOR CONTRIBUTIONSShapiro R provided expertise in the field, helped to determine article content, and aided in revisions for final product submission. Nguyen C researched agents and discussed, wrote, and reviewed the article with Dr. Shapiro.
No potential conflict of interest was reported.