



To evaluate the repair process in rats with experimentally induced arterial hypertension. This study aimed to evaluate lesions in the ventricular myocardium and the repair process during experimental hypertension induced by systemic blockage of nitric oxide using N-omega-nitro-L-arginine methyl ester hydrochloride (L-NAME). Nitric oxide is an endothelial vasorelaxing factor and is necessary for the maintenance of normal arterial pressure, and L-NAME is an analog and antagonist of L-arginine, the substrate of the nitric oxide synthase.
MATERIALS AND METHODSWe used 26 normotensive young male Wistar rats belonging to several litters. Animals were treated with oral administration of L-NAME dissolved in water (75 mg/100 mL) for 43 days. Hearts were weighed and processed by routine methods. Special stains utilized were Gomori's trichrome (aniline blue), picrosirius red polarization to identify fibrillar collagen, alcian blue technique (pH 0.5 and pH 2.5) to identify glycosaminoglycans, periodic acid-Schiff technique (with and without amylases) to identify proteoglycans, and Weigert's resorcinol fuchsin solution (with and without oxone) to identify elastic fibers.
RESULTSThe results showed significant elevation of the arterial pressure (P <0.01) and significant increase of cardiac weight (P <.0001) in the L-NAME (hypertensive) treated group, as compared to an untreated control group. The histological analysis demonstrated wide infarcted myocardial areas in animals with nitric oxide blockade; several vascular changes such as thickening of the muscular tunica with fibrosis; thickening in the wall of small arteries and arterioles; and fibrinoid necrosis in the wall to nearly complete luminal obliteration. Reparative fibrosis involved mainly oxytalan elastic and collagen fibers.
CONCLUSIONOxytalan elastic and collagen fibers are of great importance for the postinfarct repair process occurring during experimental nitric oxide inhibition.
Avaliar o processo de reparo em ratos submetidos à hipertensão arterial experimental. Avaliamos lesões miocárdicas ventriculares e processos de reparo durante a hipertensão arterial experimental induzida pelo bloqueio da síntese do óxido nítrico (ON) utilizando o Hidrocloreto de L-NAME. ON é um fator de relaxamento endotelial e é necessário para a manutenção da pressão arterial normal. L-NAME é um analogo e antagonista da L-Arginine que é o substrato da enzima óxido nítrico sintase.
MATERIAL E MÉTODOSUtilizamos 26 ratos Wistar normotensos jovens oriundos de diversas ninhadas. Os animais foram submetidos ao L-NAME (Hidrocloreto de Nwnitro-L-arginina metil ester) por administração via oral em água durante 43 dias (concentração: 75 mg / 100 ml). Os corações foram pesados e processados por métodos de rotina. Colorações Especiais utilizadas foram Tricrômico de Gomori (azul de anilina), Picro-Sirious Red sob luz polarizada para identificar as fibras colágenas, Técnica do Alcian Blue (pH 0,5 e pH 2,5) para identificar as glicosaminoglicanas, Técnica do Ácido Periódico Schiff (com e sem amilase) para identificar as proteoglicanas e Resorcina Fucsina de Weigert's (com e sem oxona) para identificar as fibras elásticas.
RESULTADOSOs resultados demonstram elevação significativa da pressão arterial (p<0,01) e significativo aumento do peso cardíaco (p<0,001) no grupo tratado com L-NAME (hipertenso), comparado com um grupo de controles não tratados. As análises histológicas demonstraram nos animais submetidos ao bloqueio da síntese do óxido nítrico amplas áreas de infarto no miocárdio, várias anormalidades vasculares como espessamento da túnica muscular com fibrose, espessamento da parede de pequenas artérias e arteríolas e, ainda, necrose fibrinóide levando à completa obliteração luminal. A fibrose reparativa teve principal presença de fibras elásticas oxitalânicas e fibras colágenas.
CONCLUSÃOAs fibras elásticas oxitalânicas e fibras colágenas são de grande importância para o processo de reparo pós-infarto que ocorre na inibição experimental do óxido nítrico.
Hypertension is a common disorder in modern industrial countries.1 In men, arterial hypertension is the most important risk factor in cardiovascular disease, affecting 11% to 20% of the Brazilian population over 20 years of age. In Brazil, cardiovascular diseases are the number-one cause of mortality, representing about 30.8% of the total deaths.2 About 90% to 95% of arterial hypertension is idiopathic, and the remaining 5% to 10% is largely secondary to renal disease. About 5% of hypertensive patients experience sharp blood pressure elevation, which if untreated, leads to death in 1 or 2 years.3 Arterial hypertension is the most important etiologic factor associated with the appearance of symptomatic heart failure, and left ventricular hypertrophy is the major risk factor.4 Ventricular hypertrophy also occurs in association with various forms of arrhythmia and is a risk factor in cardiac sudden death.5
Hypertension is a hemodynamic disorder characterized by elevation of the arterial pressure, and increased vascular resistance is its hemodynamic hallmark.6 Control of blood pressure is complex; it is determined by the interaction of multiple genetic and environmental factors.3 Even if some authors disagree,7 it is now well established that reduced activity of nitric oxide synthase is, at least in some cases, a pathogenic factor in development of hypertension.8–11
Nitric oxide (NO) is an endothelial vasorelaxing factor.12–17 N-omega-nitro-L-arginine-methyl ester (L-NAME) is an analog and antagonist of L-arginine, the substrate of nitric oxide synthase. Nitric oxide synthesis is necessary for the maintenance of normal arterial pressure, and its blockage causes arterial hypertension, significant experimental cardiac hypertrophy, and lesions in the myocardium.18–27 The extent of the direct influence of NO and the influence of hypertension produced by NO blockage on hypertrophy and on cardiac lesions is at present the subject of controversy and is subject to study using the L-NAME model. Reduction of arterial hypertension in this model does not significantly reduce cardiac hypertrophy or the lesions brought about by an NO deficit, these morbid processes are probable mainly related to the NO deficit, not the hypertension.25–26,28
The purpose of this work was to study lesions in ventricular myocardium mainly concerning oxytalan elastic and collagen fibers of rats at day 43 of arterial hypertension induced by NO synthetase blockage (L-NAME model).
MATERIALS AND METHODSTwenty-nine young normotensive adults Wistar rats from several breeds with initial weights ranging from 179 to 279 grams were used. Arterial pressure was obtained on the 40th day of the experiment using pletismography.20,27,29 To ensure that animals were normotensive, arterial pressure was measured a day before the beginning of the experiment. After arterial pressures and weights were measured, animals were placed in plastic boxes with no more than 5 to 6 animals per box. The initial arterial pressure was not above 117 mm Hg in any animal (Table 1). Seventeen rats were treated for 43 days with L-NAME (N-omega-nitro-L-arginine methyl ester hydrochloride, Sigma Chemical, St. Louis) via oral administration at a concentration of 75 mg/100 mL in drinking water (about 60 mg/kg of body weight).20,27 Nine rats were used as an untreated control group. Six hypertensive animals died during the experiment, the majority at days 41 to 43.
At day 43 of the experiment, each animal was anesthetized with ethyl ether, weighed, the thorax opened, and cardiotomy performed. The hearts were weighed, fixed in Bouin's fixative for 48 hours, processed using routine methods (embedded in Paraplast), and sliced into 6 or 7 mm sections. Sections were stained with Gomori's trichrome, using aniline blue instead of fast green; picrosirius red, by which fibrillar collagen (probable type I) takes on a red or yellow color and reticular collagen (probable type III) takes on a green color under polarized light; alcian blue technique at pH 0.5 to differentiate sulfated glycosaminoglycans and at pH 2.5 to differentiate carboxylated and phosphated glycosaminoglycans; periodic acid-Schiff technique with and without amylase to differentiate proteoglycans; and Weigert's resorcinol fuchsin solution with and without oxone. The periodic acid-Schiff technique with amylase is important for differentiating proteoglycans because it removes glycogen from the tissue.30–31 The treatment with Weigert's resorcinol fuchsin solution using oxone is important because the oxone produces oxidation, which reveals the oxytalan fibers.
RESULTSOur results demonstrate a sharp elevation of arterial pressure in rats treated with L-NAME (hypertensives) compared to the control group (Table 1). The mean tail-cuff pressure in hypertensive rats before cardiotomy (at day 40) was 176 mm Hg (range, 160 to -198 mm Hg). The control group had a mean value of 116 mm Hg before cardiomiotomy (range 100 to -123 mm Hg). Additionally, at day 40, there was a significant difference in mean cardiac weight (P <.001) between control (0.86 g, standard error = 0.04) and treatment (1.25 g, standard error = .005) groups. The ratio of cardiac weight (mg):body weight (g) was significantly different between groups (P <.001), with values of 3.30 for the control and 5.38 for the hypertensive group.
Figures 1 to 3 show examples of the histological results of the control and hypertensive groups. The histological analysis revealed that intense reactive and reparative fibrosis occurred in all layers of myocardium (subepicardial, subendocardial, and inner myocardium) in left and right ventricles of treated animals. The right ventricles were more affected. These results suggested that wide infarcted areas had developed in the myocardium. We also documented several vascular changes, such as thickening of the muscular tunica with fibrosis replacing part of the cells, narrowing of the lumina, and perivascular fibrosis sometimes showing dense collagen fibers. We observed systematically a marked thickening in the wall of small arteries and arterioles of the hypertense animals and fibrinoid necrosis in the wall to nearly complete luminal obliteration (Figures 1 and 2). In the fibrotic areas, we found the fibrillar collagen (probable type I) occupying a great proportion of total collagen. Occurrence of fibrillar collagen in reticular disposition (probable type III) was less significant. In spite of the intense fibrosis found in the hypertensive animals, the ratio of fibrillar to reticular collagens in these fibrotic areas was similar to that of the myocardial interstitium of the control group (Figure 2). Weigert's staining for elastic fibers was highly positive in the majority of the fibrotic areas, but in perivascular fibrotic areas reactivity was nil for all elastic fibers (Figure 3). Weigert's staining with oxone was more intense than that without oxone. Alterations in the concentration of the sulfated glycosaminoglycans or carboxylated or phosphated glycosaminoglycans in the myocardial mass using the alcian blue technique could not be assessed because there was no reaction.
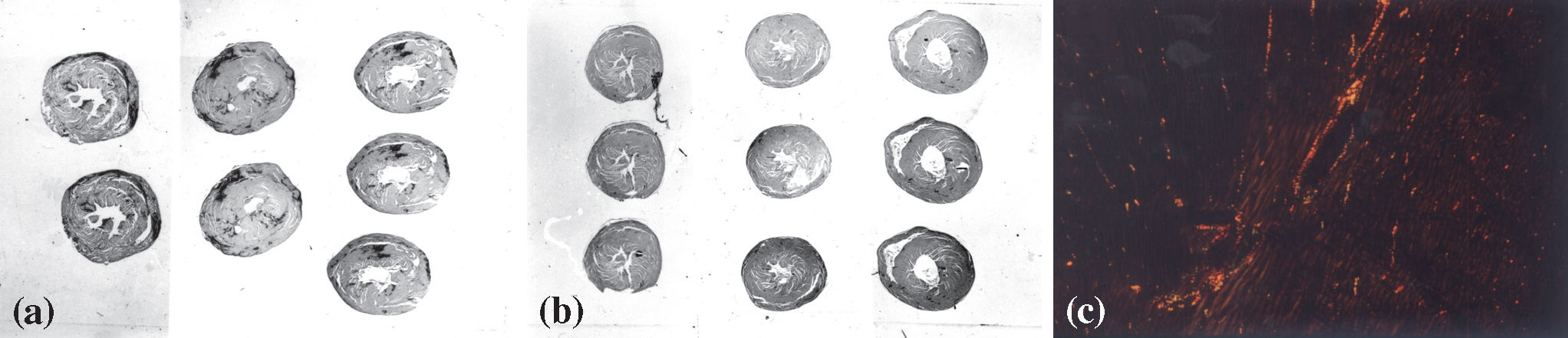
 in the hypertensive hearts (a) compared with control hearts (b) stained with picro-sirius red; (c) myocardium of a control animal showing fibrillar collagen (in yellow, orange, or red) corresponding to a high proportion of collagen, with a small amount of reticular collagen (in green) (magnification 100X) (picro-sirius red polarization).")
Panoramic view of heart sections showing hypertrophy and fibrotic scars (dark areas) in the hypertensive hearts (a) compared with control hearts (b) stained with picro-sirius red; (c) myocardium of a control animal showing fibrillar collagen (in yellow, orange, or red) corresponding to a high proportion of collagen, with a small amount of reticular collagen (in green) (magnification 100X) (picro-sirius red polarization).
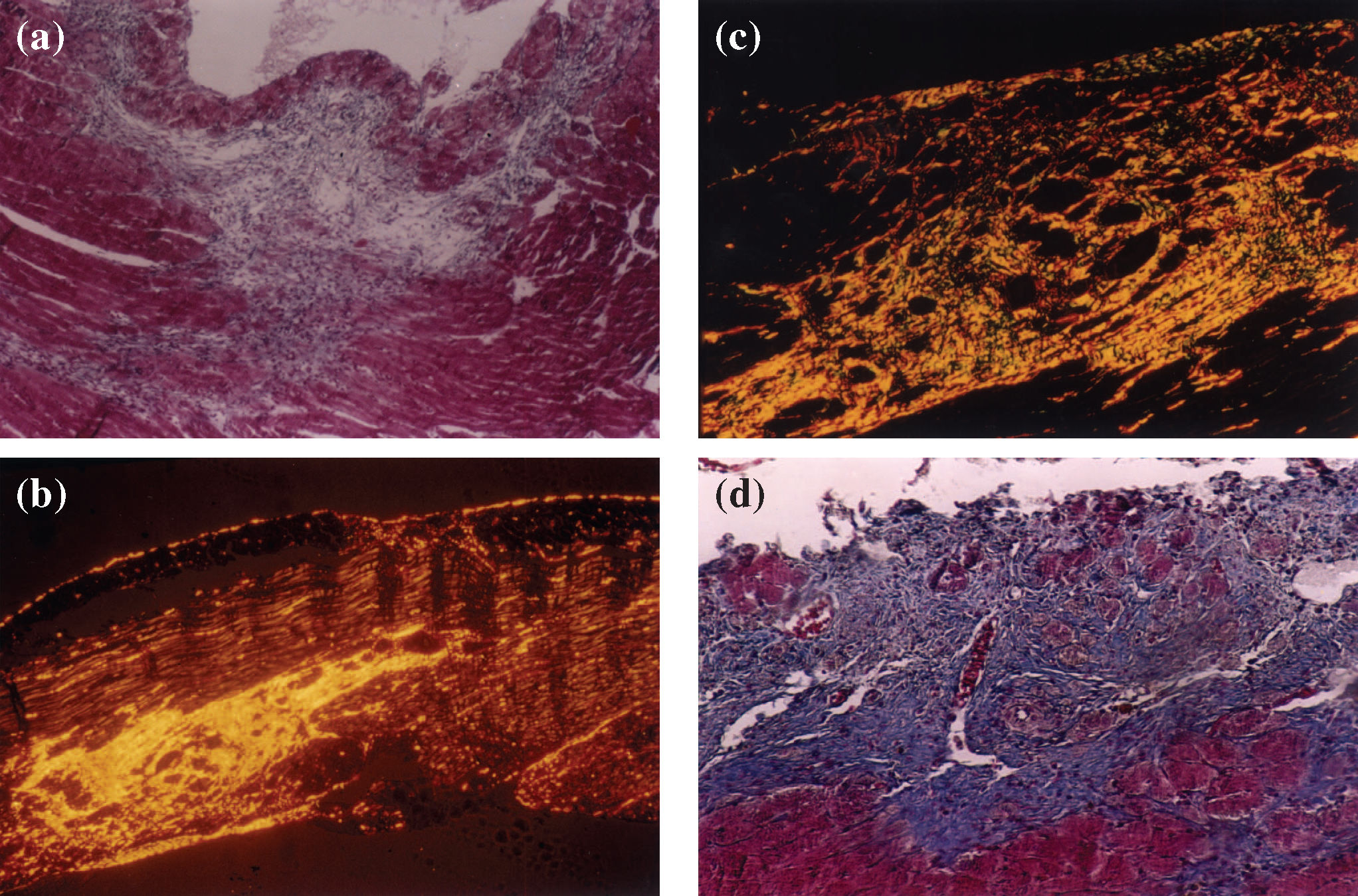
 inflammatory infiltrate areas highly suggestive of a myocardial infarct in the subendocardial region of a hypertensive heart (magnification 100X); (b) postinfarct fibrotic areas in the right ventricular myocardium of a hypertensive animal, stained with picrosirius red using polarized light to show fibrillar collagen (in yellow, orange, or red) occupying a great proportion of collagen fibers (magnification 200X); (c) postinfarct fibrotic areas in the myocardium of a hypertensive animal, showing fibrillar collagen (in yellow, orange, or red) corresponding to a high proportion of collagen, with a small amount of reticular collagen (in green) (magnification 200X) (picro-sirius-red polarization).")
(a) inflammatory infiltrate areas highly suggestive of a myocardial infarct in the subendocardial region of a hypertensive heart (magnification 100X); (b) postinfarct fibrotic areas in the right ventricular myocardium of a hypertensive animal, stained with picrosirius red using polarized light to show fibrillar collagen (in yellow, orange, or red) occupying a great proportion of collagen fibers (magnification 200X); (c) postinfarct fibrotic areas in the myocardium of a hypertensive animal, showing fibrillar collagen (in yellow, orange, or red) corresponding to a high proportion of collagen, with a small amount of reticular collagen (in green) (magnification 200X) (picro-sirius-red polarization).
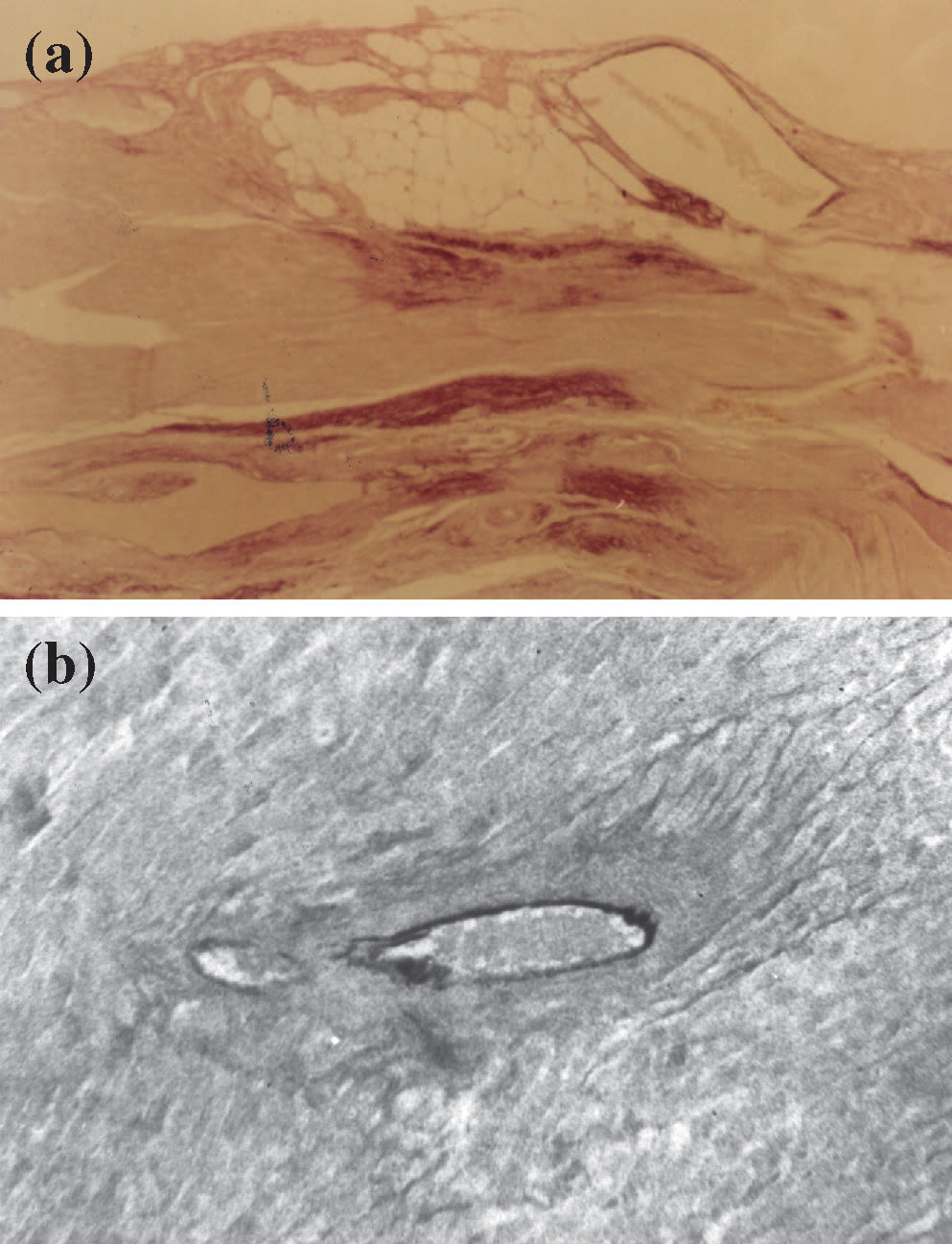
 Myocardium with intense reaction by oxytalan fibers (dark dots) of the elastic system in reparative fibrotic areas (Weigert")
(a) Myocardium with intense reaction by oxytalan fibers (dark dots) of the elastic system in reparative fibrotic areas (Weigert's resorcinol-fuchsin method using an oxone reaction; magnification 100X); (b) perivascular fibrosis characteristic of the hypertensive cardiac processes using the L-NAME model. Note the degeneration of the inner elastic lamina and lack of reaction for oxytalan fibers of the elastic system (Weigert's resorcinol-fuchsin solution with oxone; magnification 200X).
We did not find fibrotic areas in the myocardium in the control animals. These animals had vessels with characteristic normal appearance.
DISCUSSIONNitric oxide (NO) is involved in several important signal processes in cardiovascular, nervous, and immunologic systems and also functions as a modulator of extracellular matrix components.15,32–37 Nitric Oxide is produced by cardiac myocytes38 and also acts in the myocardium as a relaxing factor.22 Under physiological conditions, NO plays an important role in regulation of tissue perfusion and myocardial blood flow.39 The interruption of NO synthesis by L-NAME causes systemic arterial hypertension and significant cardiac hypertrophy and lesions (reactive and reparative fibrosis) in the myocardium.20,21,25–28 This experimental arterial hypertension can be reversed using captopril, losartan, hydralazine, and enalapril.4,20,23,25,28 The administration of L-arginine (the NO synthase substrate) in rodents treated with L-NAME reverses this experimental arterial hypertension;19 in some cases of human hypertension, the administration of L-arginine reverses the hypertensive disease.9,11
Fibrosis occurs in the myocardium during myocardial hypertrophy due to arterial hypertension. Fibrosis may occur between the cardiac myocytes (reactive or interstitial fibrosis) or secondary to myocyte necrosis (reparative or parenchymal fibrosis, or, scarring). After about 4 weeks of renovascular hypertension, reactive fibrosis occurs, and at 12 weeks, reparative fibrosis appears predominantly in endomyocardium of rats. In diastolic dysfunction (because of a pathological increase of myocardial stiffness), its major determinant is the tissue component with highest tensile strength, which is the collagen matrix of the myocardium. Type I collagen, which is predominant in the myocardium–in rats and monkeys about 70% and 82%, respectively, followed by type III at about 20% and 11%–has great tensile strength, and its concentration determines the stiffness of myocardial tissue. Thus, the origin of heart failure in arterial hypertension with diastolic dysfunction of the left ventricle appears to be rooted in the structural remodeling of the myocardium, particularly in the development of myocardial fibrosis.4 As the infarcted area heals, the late and persistent arrhythmogenic potential that emerges is not primarily dependent on the effects of ischemia on cellular electrophysiology but rather on the alteration of the tissue structure stability originating in the postinfarct reparative fibrosis.40
The ideal repair mechanism is not found in adult tissue, but rather in embryonic tissue. Because of its relative easy and ideal healing – rapid, efficient, and perfect without leaving a scar – embryonic tissue is studied as a favorite paradigm of efficient tissue repair. Identifying how embryonic tissue heals will lead to new therapeutic strategies for improving adult wound healing.41
Ribeiro et al.,20 observed that rats treated with L-NAME presented a progressive significant increase of the tail-cuff arterial pressure (168 mm Hg at day 30). They also observed systematically a marked thickening in the wall of small arteries and arterioles. They found fibrinoid necrosis in the wall of the renal arterioles and nearly complete luminal obliteration in 27% of the cases. Fibrinoid necrosis, which may result in small infarct areas, is the primary histopathological indication of malignant arterial hypertension.42
In infarcted areas after experimental coronary occlusion in rabbits, there was a 10-fold increase in the inducible form nitric oxide synthase activity.43
In the present work with the L-NAME rat model, we confirm previously reported data regarding a sharp and significant increase of the arterial pressure. We found both reactive and reparative fibrosis (including perivascular fibrosis). We also verified several vascular changes, such as thickening of the muscular tunica with fibrosis replacing part of the cells, narrowing of the lumina, and perivascular fibrosis, sometimes with dense collagen fibers. Weigert's staining both with and without oxone oxidation was highly positive in the majority of the fibrotic areas. Nevertheless, in perivascular fibrotic areas, the reactivity for Weigert's fibers was low and concerning oxytalan fibers was nil, probably involved with a lack of major stiffness of the vessels.
The oxone reaction has a high reactivity for oxytalan fibers. Weigert's resorcinol fuchsin method using oxone oxidation is used to identify oxytalan fibers, staining them violet. The Weigert's resorcinol fuchsin method without oxone oxidation is used to identify the other fibers of the elastic system.44–45 The occurrence of oxytalan fibers serves to explain at least in part the stiffness of the tissue during diastolic function, because oxytalan fibers are less distensible than the other fibers of the elastic system.46
We systematically observed a marked thickening in the wall of small arteries and arterioles of the L-NAME treated hypertense animals and fibrinoid necrosis in the wall nearly to complete luminal obliteration. We also suggest that myocardial lesions occurred by vasoconstriction (mainly of arterioles) and by the hypertrophic condition of myocytes leading to local hypoperfusion and ischemia as well as possibly by a degenerative processes of segments of the vascular bed in affected regions because of the obstructions. Concerning myocardial lesions that occur because of vasoconstriction, it is well known that in some cases, infarction occurs after vascular spasm.3 Reactive fibrosis (perivascular and perimyocyte) possibly occurs by simple interruption of NO production that would induce collagen proliferation.34–35, 38
In this work, we identified in the heart of hypertensive animals intense perivascular fibrosis and scarring of the vascular wall in the affected vascular bed. In the development of cardiac hypertrophy (along the cardiac interstitial remodeling) an abnormal accumulation of fibrillar collagen with thickening of adventitia in the intramyocardial coronaries also occurs.47 The analysis of Fischer et al.,1 following the inducement of arterial hypertension using stenosis of the left renal artery, an interstitial and perivascular focal fibrosis in the heart of hypertensive animals was found. In our material, reparative fibrosis was predominant, and we found a discreet cellular inflammatory infiltration in the lesions. This occurred because we studied animals at day 43 of administration of L-NAME, and therefore old reparative areas were present. Lukic et al.,48 studying diabetes mellitus in a mouse model, found mild inflammatory infiltration, mainly concerning some cellular types and indicated that NO played an important role in these processes. Considering all factors implicit in the origin of hypertensive disease in men, at least in some cases, the reduction of NO levels is significantly involved.11,49–51 Our present results are also similar to the abnormalities and lesions occurring, at least in some cases, in hypertension, cardiomyopathies, and in arteriosclerosis.
In the present work, we found cardiac hypertrophy at day 40 by comparing the mean of cardiac weight of the control group with cardiac weight of the hypertensive group. There was a significant difference in the cardiac weight (mg)/ body weight (g) ratio, with values of 3.30 for the control group and 5.38 for the hypertensive group. Hypertension is the most important etiologic factor associated with the appearance of symptomatic heart failure, and left ventricular hypertrophy is the major risk factor.4 Hypertrophic heart growth is in most cases considered to be induced by hemodynamic overload. However, the degree of hypertrophy is variable and is poorly correlated with the degree of arterial pressure, and is usually concentric. In some patients, the heart may double its weight; however, in others with the same arterial overload pressure - even after several years - the heart may undergo only a slight hypertrophy.42 The hypertrophic process that occurs in the L-NAME model is in some cases poorly correlated with the arterial pressure level27,49–54; however, when hypertrophy occurs, it is generally concentric.49 The diastolic dysfunction of the heart usually decreases when hypertrophy is reverted by administration of angiotensin-converting-enzyme inhibitors or calcium-channel antagonists.55 Some authors propose that this occurs not only because of the reduction of hypertrophy, but also because of the ability of antihypertensives to reduce the collagen concentration.47 The use of hydralazine or alpha-methyldopa to stimulate the reduction of hypertrophy has been hypothesized to increase fibrotic tissue.55 Our studies with hydralazine did not confirm this data.50 Some drugs, such as the angiotensin receptor antagonist, losartan;20 the calcium channel blocker, verapamil;21 the vasodilator with some mecanism mainly as a calcium channel blocker, hydralazine25; and the angiotensin-converting-enzyme inhibitors, captopril,23 enalapril,28 and trandolapril,21 revert increased arterial pressure induced by L-NAME. In our laboratory we have used hydralazine (Sigma Chemical, St. Louis) at a concentration of 120 mg/L25 as well as the commercial preparation (tablet, Ciba-Geigy) at a concentration of 36 mg/L and 120 mg/L. The dose of 120 mg/L reverted arterial pressure and postponed the abnormalities that occur during systemic nitric oxide blockage by L-NAME.50 During renovascular hypertension, pretreatment using captopril has been suggested to prevent hypertension, fibrosis, and hypertrophy of the left ventricle.4 An important question is whether the regression of myocardial hypertrophy is linked to the reduction of cardiovascular risk induced by hypertrophy.55 Nevertheless, the cardiovascular risk may also increase in some cases, for instance, when the hypertrophy is produced by a myocardial infarct.6 Vasodilators and diuretics reduce arterial pressure, however, they are not believed to reduce the cardiac weight significantly. It also has been proposed that regression of hypertrophy is only reached when sympathetic activity is reduced, because the sympathetic system is one of the crucial factors for the development of the hypertrophy.55
The author acknowledges Professors Doctor Maria de Fátima Maron Ramos, Doctor Doyle Maia, and Doctor José Vanderli Andreata. This work is part of the M.Sc. Thesis from the author, HUAP – UFF (Brazil). During this work, the author had a M.Sc. fellowship from CAPES - Brasília / Brazil. This work was supported by CNPq, CAPES, USU, and AXV.