





Pulmonary surfactant is a substance composed of a lipoprotein complex that is essential to pulmonary function. Pulmonary surfactant proteins play an important role in the structure, function, and metabolism of surfactant; 4 specific surfactant proteins have been identified: surfactant proteins-A, surfactant proteins-B, surfactant proteins-C, and surfactant proteins-D. Clinical, epidemiological, and biochemical evidence suggests that the etiology of respiratory distress syndrome is multifactorial with a significant genetic component. There are reports about polymorphisms and mutations on the surfactant protein genes, especially surfactant proteins-B, that may be associated with respiratory distress syndrome, acute respiratory distress syndrome, and congenital alveolar proteinosis. Individual differences regarding respiratory distress syndrome and acute respiratory distress syndrome as well as patient response to therapy might reflect phenotypic diversity due to genetic variation, in part. The study of the differences between the allelic variants of the surfactant protein genes can contribute to the understanding of individual susceptibility to the development of several pulmonary diseases. The identification of the polymorphisms and mutations that are indeed important for the pathogenesis of the diseases related to surfactant protein dysfunction, leading to the possibility of genotyping individuals at increased risk, constitutes a new research field. In the future, findings in these endeavors may enable more effective genetic counseling as well as the development of prophylactic and therapeutic strategies that would provide a real impact on the management of newborns with respiratory distress syndrome and other pulmonary diseases.
O surfactante pulmonar é uma substância composta por um complexo lipoprotéico essencial para a função pulmonar normal. As proteínas do surfactante têm importante papel na estrutura, função e metabolismo do surfactante. São descritas quatro proteínas específicas denominadas surfactante pulmonar-A, surfactante pulmonar-B, surfactante pulmonar-C e surfactante pulmonar-D. Evidências clínicas, epidemiológicas e bioquímicas sugerem que a etiologia da síndrome do desconforto respiratório é multifatorial com um componente genético significativo. Existem na literatura algumas descrições sobre a presença de polimorfismos e mutações em genes dos componentes do surfactante, particularmente no gene da surfactante pulmonar-B, os quais parecem estar associados à síndrome do desconforto respiratório, síndrome da angustia respiratória aguda e proteinose alveolar congênita. Diferenças individuais relacionadas à síndrome do desconforto respiratórioe síndrome da angustia respiratória aguda e à resposta dos pacientes ao tratamento podem refletir diversidade fenotípica, devido, parcialmente, à variação genética. O estudo das diferenças entre as variantes alélicas dos genes das proteínas do surfactante pode ajudar na compreensão das variabilidades individuais na susceptibilidade ao desenvolvimento de várias doenças pulmonares. A determinação de quais polimorfismos e mutações são, de fato, importantes na patogênese das doenças relacionadas à disfunção das proteínas do surfactante e a possibilidade da realização da genotipagem em indivíduos de alto risco constitui um novo campo de pesquisa, que pode permitir, futuramente, um aconselhamento genético mais efetivo, resultando no desenvolvimento de estratégias profiláticas e terapêuticas que representem um impacto real no manejo dos recém-nascidos portadores da síndrome do desconforto respiratório e outras patologias pulmonares.
Pulmonary surfactant is a substance composed of a lipoprotein complex that is essential to normal pulmonary function; it is responsible for the decrease in superficial tension of the alveolar air-liquid interface, therefore preventing pulmonary collapse on expiration.1
In 1959, Avery and Mead suggested that surfactant deficiency could cause hyaline membrane disease, currently called respiratory distress syndrome (RDS).1 The inability of the preterm newborns to produce adequate quantities of surfactant due to pulmonary immaturity constitutes the primary etiology of respiratory distress syndrome (RDS).2
Decreased function of the surfactant also contributes to the physiopathology of acute respiratory distress syndrome (ARDS),3 characterized by acute pulmonary lesion with pulmonary edema, nonhydrostatic and severe hypoxemia, and high mortality rates of 10% and 90% (average, 50%), depending on the etiological factor.
Respiratory distress syndrome constitutes one of the main causes of respiratory morbidity and mortality in children under 1 year of age.4 Despite the improvement in neonatal survival rates, a high percentage (5%-25%) of newborns affected by RDS will develop chronic pulmonary disease.
High pulmonary morbidity has been attributed to toxicity by oxygen, barotrauma, immaturity in development, and nutritional deficiencies in these newborns. However, significant differences in the evolution of the pulmonary condition of newborn carriers of RDS having similar clinical characteristics and comparable risk factors (for example, the need for oxygen and mechanical ventilation), suggest that other factors, such as genetics, can also contribute to these different clinical evolutions.5,6
The surfactant systemThe surfactant complex comprises lipids (90%) and proteins (10%). Among the lipids, about 70% is phosphatidylcholine predominating as dipalmitoylphosphatidylcholine. The second most important phospholipid component is phosphatidylglycerol (10%); other phospholipids are phosphatidylinositol, phosphatidylethanolamine, and phosphatidylserine (Figure 1).

Composition of surfactant. Modified from Jobe et al10
The surfactant proteins (SP) play an important role in the structure, function, and metabolism of the surfactant. Four specific proteins have so far been described: SP-A, SP-B, SP-C, and SP-D. These proteins are synthesized and secreted by type II epithelial cells and are classified into 2 groups: the hydrophilic (SP-A and SP-D) and the hydrophobic (SP-B and SP-C).7 Although the protein concentration of surfactant is significantly less than that of the phospholipids, the surfactant proteins are fundamentally important for the prevention of alveolar collapse.8 The surfactant proteins are already well characterized regarding the genome, amino acid composition, and messenger RNA (mRNA) sequence.9
There are two functional genes for SP-A (SP-A1 and SP-A2) and one pseudogene,11 all being necessary for a well-functioning, mature, and stable SP-A.12 The gene responsible for its production is located on the long arm of chromosome 10.13 It is the most abundant protein, comprising 5% of the surfactant composition.14 SP-A is hydrophilic, a member of the collectin family, and presents recognition domains for collagen and carbohydrates that relate to pulmonary defense and pulmonary inflammatory processes.15 The de novo synthesis of SP-A occurs in a continuous and independent manner from the lamellar bodies and other surfactant components.16 Once in the aerial space, SP-A associates with lipids, forming tubular myelin, a structural form of the surfactant. In the mature lung, SP-A is expressed predominantly in type II epithelial cells and Clara cells, and it is also expressed in tracheal gland cells.10
SP-B is a hydrophobic protein and constitutes 2% of the surfactant composition.17 The locus of the gene responsible for the production of SP-B is on the short arm of the chromosome 2 (2p12→p11.2), extends for approximately 9.5 kilobases, and contains 11 exons.18 It is secreted by type II epit helial cells and is essential for pulmonary function, since it promotes the adsorption and distribution of the phospholipids on a single layer, which corresponds to the functional form of the surfactant.19 SP-B and SP-A are essential for the formation in vitro of the tubular myelin, the morphologic form of the surfactant. A well-structured tubular myelin was found to be absent in newborns who died due to RDS.20
Deficiency in SP-B is associated with an alteration of production in normal lamellar bodies, blocking surfactant secretion—not to the processing of active SP-C—leading to a progressive respiratory insufficiency that is incompatible with life.21
SP-C is a hydrophobic protein, constituting about 1% of the surfactant composition.22 SP-C is expressed in type II epithelial cells, processed in multivesicular bodies to its mature form, stored in the lamellar bodies, and secreted with SP-B and lipids.17 It has similar functions to those of SP-B, ie, promoting the adsorption of phospholipids. Contrary to the case with SP-B, mice that do not produce SPC process SP-B normally and survive with no apparent pulmonary disease.23 However, in humans, SP-C deficiency can lead to pulmonary interstitial disease on the first years of life. Experimental models demonstrate that tumor necrosis factor-alpha and viral infections lead to the decrease of SP-C mRNA, which seems to lower the surfactant function.25,26
SP-D is a hydrophilic protein and has structural and functioning similarities with SP-A. It constitutes 1% of the surfactant composition and is a member of the collectin family. In the lungs, SP-D is expressed by type II epithelial cells, Clara cells, and other cells and glands. The major part of SP-D is not associated with lipids.10 SP-D is related to pulmonary defense, but contrary to the case with SP-A, it increases in the presence of acute pulmonary lesion. Rats that do not produce SP-D possess an increased pool of lipids in the tissues and alveolus without an increase of the surfactant proteins; these rats develop emphysema.27,28 Congenital SP-D deficiency has so far not been reported in humans.
Surfactant metabolismDipalmitoylphosphatidylcholine is synthesized on the rough endoplasmic reticulum and transferred to the lamellar bodies together with SP-B and SP-C (Figure 2). The lamellar bodies are the storage and secreting granules and are surrounded by a limiting membrane that fuses with the plasma membrane. Surfactant secretion can be stimulated by the stretching of the type II epithelial cells, by the action of beta-agonists, and purinergic agonists, such as ATP.29

Surfactant intracellular metabolism. Modified from Whitsett et al30
After secretion to the interior of the alveolus, the surfactant goes through a complex cycle. The fat and protein molecules form the tubular myelin. With the successive movements of contraction and stretching that occur at each respiratory cycle, part of the myelin becomes disorganized and disassociates from the main film, forming small vesicles that are reabsorbed to the interior of type II epithelial cells. Inside the cell, a small portion is catabolized, whereas most of the reabsorbed surfactant is reorganized on the lamellar bodies in a recycling process. In preterm newborns, 50% of the alveolar pool is composed of inactive vesicles that will be recycled. This relationship is more unfavorable in conditions of pulmonary lesion, such as in RDS cases.10
Surfactant proteins: genetic determinants and pulmonary diseasesThe importance of SP-B for the function of the surfactant is clearly exemplified by the results of studies using “knock-out” mice for the SP-B gene, that is, mice in which the gene responsible for the production of SP-B has been experimentally removed. Despite the fact that lungs of SP-B (-/-) mice develop normally, they are atelectatic at birth, and the mouse dies due to acute respiratory insufficiency. On the other hand, heterozygous mice (+/-) do not present early respiratory insufficiency.31 Heterozygous mice present reduced pulmonary compliance and increased pulmonary residual volume; therefore, their lungs are vulnerable to pulmonary lesion, and they develop respiratory failure when submitted to hyperoxia.32,33
Some descriptions about the presence of polymorphisms and mutations in genes from the surfactant components are reported in the literature, particularly regarding the SP-B gene, which seems to be associated with RDS, ARDS, and CAP.34–40
The inability to produce SP-B is the result of an autosomic recessive disease that leads to a fatal respiratory insufficiency, congenital alveolar proteinosis (CAP), indicating the fundamental role this protein plays in normal pulmonary function.41
In 1994, Nogee et al42 demonstrated the existence of a mutation-type frameshift, consisting of the substitution of a nucleotide sequence, GAA, with a single nucleotide, C, on codon 121 of the gene responsible for the production of SP-B. In the index family studied by the authors, 3 term newborn babies developed fatal respiratory insufficiency, while the parents and 3 siblings did not present any respiratory symptoms, suggesting a recessive autosomic inheritance pattern. SP-B was not detected in the pulmonary tissue of the children with the disease, and SP-B mRNA was not detected by the Northern blot method.41,42 The authors concluded that this mutation could be responsible for SP-B deficiency and for CAP, and they suggested that the disease could have a higher incidence than that reported by them previously in 1993.
In 1998Lin et al,43 identified 3 mutations (2 substitutions and 1 deletion) on the gene of protein B from the surfactant of 3 individuals with a CAP diagnosis from the same family.43 The most important was the mutation 122delT, which resulted in an anomalous and nonfunctioning SP-B; a “hot-spot” (presence of 3 mutations in a small region of the gene) was identified on exon 4 of the SP-B gene. The authors also suggested the possibility of mutations responsible for CAP or other respiratory diseases occurring in a region near the “hot-spot” described in this study.
After 2 years, Lin et al published a study of a family with a history of 14 newborn deaths due to early respiratory insufficiency.44 The immunohistochemistry of the lung from 3 of these children showed a decrease or absence of SP-B expression. Nine polymorphisms were found in this family, but it was not possible to attribute any of them to the SP-B deficiency. The analysis of the mRNA using RT-PCR (reverse transcript polymerase chain reaction) of pulmonary tissues embedded in paraffin from patients with CAP demonstrated that the mRNA of SP-B showed alterations. The authors concluded that the CAP problem in this family could reflect aberrations on the mRNA of the B protein of the surfactant. Nogee et al described 13 new mutations on the gene from SP-B responsible for SP-B deficiency, indicating a high level of allelic and biochemical heterozygosis regarding CAP.37
Newborns with hereditary SP-B deficiency are usually full-term infants who develop respiratory insufficiency during the first hours of life. The clinical condition is similar to that of preterm newborns with RDS. However, despite the treatment with surfactant, mechanical ventilation, and extracorporeal membrane oxygenation (ECMO), these babies usually die in the first months of life due to respiratory insufficiency. Immunohistochemistry shows an absence of proSP-B and SP-B, and the definitive diagnosis is achieved through DNA sequencing, showing the presence of the mutation of complementary DNA (cDNA) of the affected individuals and relatives. More than 25 distinct mutations in the SP-B gene have been identified, and all of them have a recessive autosomic genotype.37 The most frequent mutation of SP-B deficiency is 121ins2; its frequency is 1 for each 1000 to 3000 individuals.41,45
Deficiency in SP-C is associated with severe lung disease in neonates, and most mutations in the SP-C gene are inherited as a dominant gene. Various mutations on the SP-C gene have been described in infants with RDS and severe chronic lung disease, and in nonspecific interstitial pulmonary pneumonitis, interstitial pulmonary fibrosis, and interstitial fibrosis in older individuals. The age and pathology presentation are variable, and the pathological findings depend on age, clinical progression, and presence of infections.6,24,46
Recently, mutations of the transport protein ABCA3 have been associated with respiratory failure in full-term newborns, with a recessive autosomic inheritance. The protein ABCA3 is a member of the ATP-dependent transporter family, as is the cystic fibrosis transmembrane conductance regulator and multiple-drug-resistant protein known for transporting a variety of ligands, ions, proteins, peptides, carbohydrates, and lipids.47
The transporting protein ABCA3 has been identified in the membrane that limits the lamellar bodies, and it is expressed in the lung epithelial cells.48 Newborns with a deficiency of this protein develop respiratory failure during the first 24 hours of life that is refractory to the use of surfactant and ventilation. Chest radiography can detect surfactant deficiency, which is revealed by a ground-glass pattern. The pulmonary pathology is compatible with alveolar proteinosis and desquamating interstitial pneumonia.
In cases of ABCA3 deficiency, electronic microscopy shows structural alterations of small lamellar bodies, with densely colored inclusions.47 Tubular myelin is absent, and the airspaces are filled with macrophages, lipid, and protein debris. It has been suggested that ABCA3 deficiency causes abnormalities in surfactant conditioning, secretion, and function, leading to respiratory failure. It has been recently demonstrated that patients with mutations on ABCA3 present surfactant with phosphatidylcholine deficiency and altered function.49 Despite the fact that the prevalence of mutations of ABCA3 has not yet been determined, such pathology is more frequent than deficiencies of SP-B and SP-C, and it can also contribute to family lung interstitial disease in adults and children.
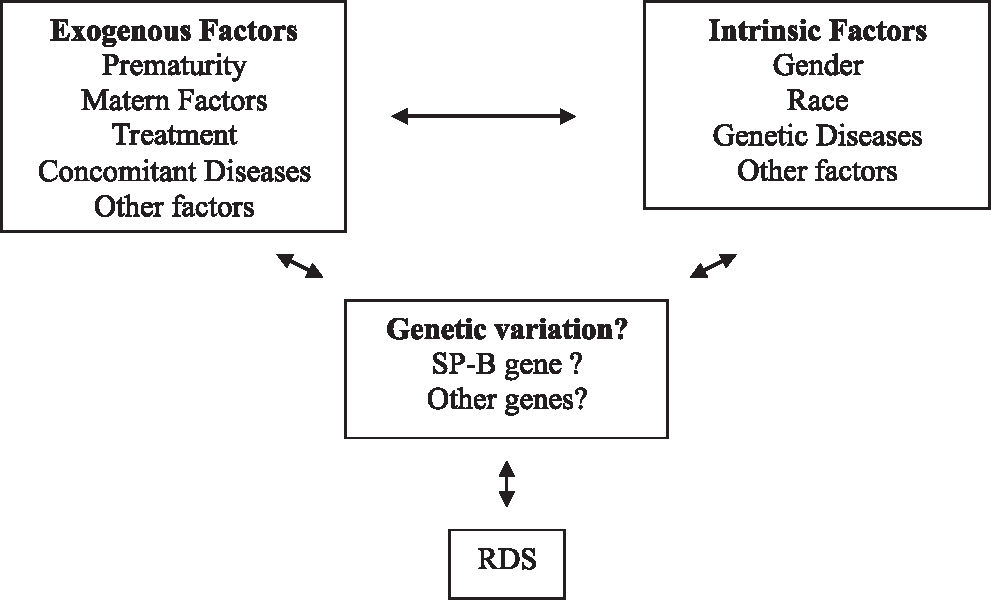
Genetic factors of respiratory distress syndromeClinical, epidemiological, and biochemical evidence suggests that the etiology of RDS is multifactorial, with a significant genetic component.6,34 Prematurity is one of the most important predisposing factors of the RDS etiology; it is rare in full-term newborns.6,34 The male gender constitutes a risk factor for the development of RDS.51,52 Black preterm newborns present a lower and less severe incidence of the disease than white newborns.51,53Figure 3 a the schematic representation of environmental and genetic factors and the interaction between these and the RDS phenotype.

Factors that contribute to the RDS phenotype. Modified from Haataja et al6
Due to the importance of the surfactant proteins on the biology and physiology of the surfactant, various authors have suggested that under certain circumstances these proteins are factors that can contribute to the etiopathogenesis of RDS.34,36 The authors have demonstrated that there is a positive synergism between one SP-A allele and a polymorphic variant of SP-B and RDS. Other authors had previously described that certain polymorphisms of SP-B occur at a high frequency in a group of patients who are RDS carriers.35
Individual differences related to RDS and the patient’s response to the treatment can reflect phenotypic diversity, due partially to genetic variation.
Some genes seem to be involved in the pathogenesis of RDS, with the genes for SP-A and SP-B being the most studied ones. This is due to the direct importance of these proteins to surfactant biology and to the development of RDS. Recently, most studies have reported the importance of the interaction between SP-A and SP-B regarding genotyping analysis in newborns with RDS. This interaction could be responsible for the determination of a higher or lower risk of developing RDS.
The SP-C gene has various polymorphic sites in different locations. Association between this gene and RDS cannot be ruled out.7,54,55 Although the knock-out mouse for the SP-C gene is viable and grows normally without significant alterations, the lungs have reduced compliance.23
Abnormalities observed on the knock-out mouse for the SP-D gene, as well as the increase in the alveolar phospholipid pool, suggest that SP-D could be more important than previously thought.27,28 However, there is no evidence so far indicating that allelic variations can be attributed or not to RDS.
The genes that codify the surfactant proteins A, B, C, and D contain various polymorphisms on their nucleotide sequences. Many of these variations are polymorphisms of a single nucleotide (single nucleotide polymorphism—SNPs), which result from substitutions of amino acids or silent modifications, whereas others are located on the introns or on 5’ UTR or 3’ UTR regions and do not cause alterations in the codified peptide sequence, but they can affect the level of transcription or posttranscription.6
The study of the differences between the allelic variants of the surfactant protein genes can help explain individual variability regarding susceptibility for the development of various pulmonary diseases. SP-B is one of the most important components of the surfactant system, and various studies have showed this relationship.
Floros et al, in 2001,56 genotyped individuals with and without RDS for variants of intron 4 and for 4 polymorphisms of SP-B: AC-18, AC-1013, CT-1580, and AG-9306. The objective of these authors was to study case-control associations in black newborns and white newborns. A possible interaction was also analyzed between the existence of specific variants of SP-B and protective SP-A variants regarding susceptibility to RDS, aiming to determine the existence of an increased or decreased risk for RDS.
The results indicate that different polymorphisms on intron 4 of the SP-B gene are associated with different subgroups of RDS in white (deletion variant) and black (insertion variant) individuals. In each racial group, the corresponding variant seemed to be a risk factor for white male and black female individuals. In white individuals, the genotypes of SP-A, 6A2, or 1A0, in the presence of a certain genotype of SP-B (9306 A/G) or intron 4 (del/*), showed an increased risk for RDS. In black individuals, however, the SP-A1, 6A3 protector genotype, in the presence of genotype SP-B 1580 (T/T), showed an increase protection against RDS, especially in individuals with advanced gestational ages.56
The polymorphism 1580 (C/T), present at the end of exon 4, on nucleotide 1580, affects the amino acid 131, altering the threonine amino acid (ACT) to isoleucine (ATT). Such an alteration eliminates a potential site of glycosylation and is associated with certain pulmonary pathologies.44 WANG et al57confirmed the existence of an N-linked glycosylation site at the terminal portion of SP-B gene. The T allele of the polymorphism 1580 (C/T) does not have the N-linked glycosylation site at the N-terminal fragment, and it is considered a protector factor regarding RDS, whereas the allelic variant C, which contains the recognition N glycosylation site of the fragment N-terminal, is a risk factor for chronic obstructive pulmonary disease (COPD) and ARDS.39,58
Martilla et al59 analyzed pairs of preterm twins of the same sex of white race, where at least one of them presented RDS, and showed that genotypes specific for SP-A and SP-B could influence the susceptibility to RDS. Regarding the protection against the development of the disease, it was evident both in the heterozygous C/T and homozygous T/T genotype. The authors suggested that the genotype T/T seem to increase the protection in relation to the disease, as was demonstrated with the black patients with RDS by Floros et al.56
In a study conducted in Finland, the genotype C/C of the polymorphism C/T 1580 seemed to constitute both a risk factor for the alleles of SP-A for susceptibility to RDS and a protective factor for the allele 6A3.60 Guo et al showed that the allele C seemed to be associated with a high risk of chronic obstructive pulmonary disease.58 Most of studies available suggest that the genotype C/C seems to be associated with a higher risk of pulmonary disease.
The polymorphism A/G on nucleotide 9306 is located on region 3’UTR, just 4 nucleotides above the signaling region TAATAAA. In vitro studies suggest that SP-A and SP-B can interact functionally in a synergic way to reduce alveolar tension.61 Both SP-A and SP-B are necessary for the formation of tubular myelin, considered the structural form of the surfactant. The levels of SP-A and SP-B and of tubular myelin are decreased or absent in the lungs of newborns with RDS.20,62,63 In a study by Kala et al, with a control group (n = 86 whites and 12 blacks) and an RDS group (n = 106 whites and 37 blacks) with gestational ages varying from 24 weeks up to term, a positive synergic association was observed between the allele 1 A0 and the polymorphic variant of the intron 4 and RDS.36
In 2001 Floros et al conducted a case-control study with preterm newborns with and without RDS.56 The authors concluded that the genotype A/G of the polymorphism A/G 9306 seems to be a susceptibility factor for RDS in individuals with gestational age greater than or equal to 33 weeks. The presence of specific alleles of SP-A and SP-B in the same individual resulted in a considerable change of the values of P or odds ratio, when compared to the values obtained with each genotype separately.
The polymorphism A/C – 18 is located on region 5’UTR, 11 nucleotides below the TATAAA box and 18 nucleotides to the left of the transcription initiation site, which could have an impact on the beginning of mRNA transcription. In the case-control study conducted by Floros et al to verify the association of SP-A and SP-B polymorphisms in white and black newborns with RDS, the authors observed that, in black individuals, the frequency of the genotype A/A of the polymorphism A/C – 18 was higher (P = 0.04) when compared to genotype A/C. This observation was found in preterm babies with RDS with gestational ages between 28 and 31 weeks. However, the number of individuals on this group was very small (control group, n = 4; RDS group, n = 19). No significant interaction was observed between either of the two loci and the alleles of SP-A.56
In 2003, Liu et al conducted a study to evaluate the similarity of genetic markers among populations from 3 different ethnic groups (Caucasians, Blacks, and Hispanics), with the objective of observing whether people from different races or ethnic groups could be grouped in linkage analysis studies. The results showed that the allele and genotype frequencies could be different among diverse ethnic groups, especially among ethnic groups of different races.64
SP-B polymorphisms and other pathologiesIn 2000 in Mexico, a group of researchers analyzed the polymorphisms of SP-A, SP-B, and SP-D genes in a sample of a Mexican population with chronic obstructive pulmonary disease and concluded that the alleles of these proteins could be useful for indicating subgroups that could benefit from clinical treatment.58
An analysis of polymorphisms for SP-A, SP-B, and SP-D genes was conducted in 52 patients with ARDS (idiopathic causes: pneumonia and others; exogenous causes: surgery, trauma, and aspiration), 25 patients at risk of developing ARDS, and 46 healthy controls. The authors concluded that the determination of polymorphisms on genes that codify the surfactant proteins could be useful for the study of pulmonary diseases, and that the polymorphism C/T1580 could serve to differentiate subgroups of patients with ARDS.39
Some studies suggest that allelic variations of SP-A and SP-D could be associated with bronchiolitis caused by the respiratory syncytial virus. The amino acids at position 223 of the SPA2 gene and 11 of SP-D gene could be involved in the susceptibility to infections by the respiratory syncytial virus, and they could be potential targets for prophylaxis and treatment with specific surfactant preparations.6,65,66
These studies indicate that the genetic variants of surfactant proteins could serve as valuable markers for the genetic mapping of various pathologies, particularly RDS. Additionally, racial differences have been described in other studies as important risk factors for RDS. Taken together, these studies support the possibility of a multifactorial and multigenic etiology for RDS.
Despite the fact that various recent reports discuss the complex etiopathogenesis of RDS from the molecular biology point of view, some questions remain unanswered, such as the fact that some premature newborns present distinct clinical evolutions regarding the development of RDS, as well as the different response of these newborn to the maternal use of prophylactic corticoids.
New studies are necessary for a better understanding of the specific mechanisms and the phenotypic consequences of protector allelic variants or those that predispose to respiratory disease.
The determination of which polymorphisms and mutations are, in fact, important in the pathogenesis of diseases related to dysfunction of the surfactant proteins, together with the possibility of conducting genotyping in high-risk individuals, constitutes a new field of research that may in the future facilitate more effective genetic counseling, and result in the development of prophylactic and therapeutic strategies that make a real impact on the management of newborns having or at risk for RDS and other pulmonary pathologies.