



To examine the association of atherogenic and thrombogenic markers and lymphotoxin-alfa gene mutations with the risk of premature coronary disease.
METHODS:This cross-sectional, case-control, age-adjusted study was conducted in 336 patients with premature coronary disease (<50 years old) and 189 healthy controls. The control subjects had normal clinical, resting, and exercise stress electrocardiographic assessments. The coronary disease group patients had either angiographically documented disease (>50% luminal reduction) or a previous myocardial infarction. The laboratory data evaluated included thrombogenic factors (fibrinogen, protein C, protein S, and antithrombin III), atherogenic factors (glucose and lipid profiles, lipoprotein(a), and apolipoproteins AI and B), and lymphotoxin-alfa mutations. Genetic variability of lymphotoxin-alfa was determined by polymerase chain reaction analysis.
RESULTS:Coronary disease patients exhibited lower concentrations of HDL-cholesterol and higher levels of glucose, lipoprotein(a), and protein S. The frequencies of AA, AG, and GG lymphotoxin-alfa mutation genotypes were 55.0%, 37.6%, and 7.4% for controls and 42.7%, 46.0%, and 11.3% for coronary disease patients (p = 0.02), respectively. Smoking, dyslipidemia, family history, and lipoprotein(a) and lymphotoxin-alfa mutations in men were independent variables associated with coronary disease. The area under the curve (C-statistic) increased from 0.779 to 0.802 (p<0.05) with the inclusion of lipoprotein(a) and lymphotoxin-alfa mutations in the set of conventional risk factors.
CONCLUSIONS:The inclusion of lipoprotein(a) and lymphotoxin-alfa mutations in the set of conventional risk factors showed an additive but small increase in the risk prediction of premature coronary disease.
Coronary atherosclerosis and its complications constitute a complex and polygenic disorder resulting from the combined effects of multiple environmental and genetic factors (1). Several traditional risk factors increase the risk of coronary heart disease (CHD) and heart attack (2-4). Coronary artery disease (CAD) results from the progression of atherosclerotic plaque. Studies show that almost half of an individual's susceptibility to CAD is heritable (3). Lipid metabolism and inflammation have been identified as main biological pathways in the pathogenesis of CAD. Arterial inflammation is a key component of plaque progression and plaque rupture, with atherosclerotic lesions established as active sites of inflammation (4). Biomarkers, coagulation factors, and proteins appear to coordinate the development of atherosclerosis and lead to the formation of complex atherosclerotic plaques and thrombus formation (5-7). Several biomarkers and cytokines likely play a role in determining the degree of inflammation and contribute to the promotion or retardation of atherothrombosis development. Despite the improved understanding of atherothrombosis pathophysiology, the conventional CAD risk factors do not fully account for the overall CAD risk (8,9). Moreover, risk factors associated with premature CAD are poorly recognized. Studies also show conflicting results with respect to some atherogenic and thrombogenic biomarkers of CAD (10-12). Genetic influence is often suggested as an important factor associated with the risk of premature CAD (13). The proinflammatory cytokine lymphotoxin-alfa (LTA) is a key mediator of the initiation of local vascular inflammatory responses. The actions of LTA are characterized by the stimulation of adhesion molecule production, thrombogenesis, smooth muscle proliferation, platelet activation, release of vasoactive agents (14-17), and control of nitric oxide production (8). Functional single nucleotide polymorphisms (SNPs) in the LTA gene have been found to be associated with MI in the Japanese population (17,18). However, other researchers failed to provide evidence of the relationship between LTA and CAD (19-21). The mutant allele results in significantly increased production of LTA in mononuclear cells stimulated in vitro, which is related to increased gene transcription (17). The aim of this study was to investigate whether traditional risk factors, thrombogenic and atherogenic biomarkers, and the mutation A252G in the LTA gene may be related to an increased predisposition to premature CAD and its interaction with traditional risk factor modulation.
MATERIALS AND METHODSStudy populationA cross-sectional, case-control, age-matched study was conducted in 525 consecutive subjects <50 years of age. Clinical characteristics and laboratory data were analyzed in 189 healthy control individuals and in 336 patients with documented premature stable CAD (<50 years of age) selected from the outpatient clinic of the Heart Institute (InCor), Faculdade de Medicina da Universidade de São Paulo, Brazil. We hypothesized that young adult patients exhibit a larger proportion of CAD driven by genetic risk factors compared with older patients with CAD. The control group with no CAD included healthy subjects with normal clinical histories, physical examinations, and resting and exercise stress electrocardiographic assessments. The CAD group included outpatients from the Heart Institute (InCor) with angiographically documented disease (>50% luminal reduction) and patients with a previous myocardial infarct episode. The local institutional review boards approved the study protocol, and written informed consent was obtained from each study participant.
Data collectionThe clinical data obtained included age, gender, body mass index (weight [kg]/height [m2]), smoking history, arterial hypertension, diabetes, dyslipidemia, previous myocardial infarction, and family history of premature CAD. Smokers who had quit more than 1 year prior to the study were considered ex-smokers. Current and former smokers were considered as one group and were compared with the patients who had never smoked. Hypertension was defined as a systolic blood pressure ≥140 mm Hg, diastolic blood pressure ≥90 mm Hg, or both measured at study entry or following antihypertensive treatment. A family history of CAD was defined as obstructive CAD occurring in parents (before age 55 for men and 65 for women) and siblings. Dyslipidemia was considered as the presence of hypercholesterolemia (total cholesterol >6.2 mmol/L) or hypertriglyceridemia (triglycerides >2.87 mmol/L) with medication use (e.g., fibrates and statins). Diabetes was diagnosed when a patient was taking hypoglycemic drugs or exhibited fasting glucose blood levels >7.0 mmol/L. No patients used contraceptives or hormone replacement therapy. Thrombogenic factors (fibrinogen, assessed by the Clauss method; protein C, protein S, and antithrombin III, assessed by chromogenic methods); and atherogenic factors (fasting glucose and lipid profiles, assessed by standard protocols; and lipoprotein (a) and apolipoprotein AI and B fractions, assessed using the immunoturbidimetric method) were determined.
GenotypingGenomic DNA was extracted from peripheral blood leukocytes by the standard salting-out procedure (10). We studied SNP 252A>G (rs909253) in the LTA gene, which encodes LTA on Chromosome 6p21 in intron 1 (11). The genetic variability of LTA was determined by amplification of the genomic DNA using polymerase chain reaction (PCR) followed by the restriction fragment length polymorphism (RFLP) technique. A 30-cycle PCR was performed in a PTC-DNA Engine Tetrad2 (MJ Research, Waltham, Massachusetts, USA) using a 10-μL reactive solution containing 10 mM Tris-HCl (pH 9.0), 50 mM KCl, 2.5 mM MgCl2, 100 μM of each dNTP, 0.3 U of Easy Taq DNA Polymerase, 5 pmol of each primer, and 1 μL of genomic DNA template. PCR products were digested with 1 U of NcoI restriction enzyme and visualized using 2.5% agarose gel electrophoresis. The presence of the 782-pb fragments indicated homozygosis of the wild allele (AA), while the presence of the 586- and 196-pb products indicated homozygosis of the mutant allele (GG) and the presence of 782-, 586-, and 196-pb products indicated heterozygosis (AG genotype). Genotype distribution was in Hardy-Weinberg equilibrium in all study samples (p>0.001).
Statistical methodsThe Hardy Weinberg equilibrium for the distribution of genotypes was estimated by the chi-square test in the groups studied (no-CAD and CAD). Chi-square tests, t tests, and analysis of variance with Tukey's correction for multiple comparisons were used for baseline comparisons. The odds ratios for different association models were calculated with a 95% confidence interval (CI) by multiple logistic regressions with confounders determined by a backward conditional elimination method for a significance level below 0.05. Logistic regression was used to estimate the cross-sectional association of the traditional risk factors for CAD with the following independent variables: gender, smoking status, family history of premature CAD, diabetes, dyslipidemia, history of arterial hypertension, Lp(a), and LTA polymorphisms. Lp(a) was dichotomized into normal (<30 mg/dL) and high (≥30 mg/dL) values. CAD was the dependent variable. To evaluate the model's performance, a receiver operating characteristic (ROC) curve was developed, and the area under the ROC curve (AUC or C-statistic) was used to measure the discriminative power. The C-statistic for the clinical model and the biomarker information added to the clinical model were compared. The significance level adopted for the statistical tests was 5% (p<0.05). Statistical analyses were performed using the SAS for Windows (Statistical Analysis System) program, version 9.2 (SAS Institute Inc., 1989-1996, Cary, NC, USA).
RESULTSBaseline characteristicsThe demographic and laboratory characteristics of the study population are shown in Table 1.
Demographic and laboratory characteristics of the study population.
| All patients N = 525 | Controls N = 189 (36%) | Coronary artery disease patients N = 336 (64%) | p-value | |
|---|---|---|---|---|
| Age (years) | 40.4±5.3 | 40.7±5.9 | 40.2±5.0 | 0.275 |
| Women (%) | 271 (51.6) | 69 (36.5) | 202 (60.1) | <0.001 |
| Hypertension | 150 (28.6) | 33 (17.5) | 117 (34.8) | <0.001 |
| Smokers | 56 (10.7) | 7 (3.7) | 49 (14.6) | <0.001 |
| Diabetes | 87 (16.6) | 23 (12.2) | 64 (19.2) | 0.039 |
| Dyslipidemia | 298 (56.9) | 83 (43.9) | 215 (64.2) | <0.001 |
| Family history of CAD | 255 (48.8) | 33 (17.5) | 222 (66.5) | <0.001 |
| Body mass index (kg/m2) | 25.4±5.8 | 24.9±5.4 | 25.6±6.0 | 0.235 |
| Leukocytes (mm3) | 6647±1747 | 6467±1632 | 6753±1806 | 0.080 |
| Platelets (mm3) | 181,730±112,077 | 178,784±102,551 | 183378±117185 | 0.641 |
| Cholesterol (mg/dL) | 115.5±64.2 | 114.0±49.4 | 116.4±71.3 | 0.656 |
| HDL-cholesterol (mg/dL) | 42.1±13.3 | 45.7±13.0 | 40.0±13.0 | <0.001 |
| LDL-cholesterol (mg/dL) | 69.3±34.4 | 65.7±27.4 | 71.5±37.8 | 0.098 |
| Triglycerides (mg/dL) | 127.5±213.0 | 106.6±122.4 | 138.8±248 | 0.078 |
| Glucose (mg/dL) | 96.1±32.3 | 91.0±11.2 | 99.2±39.7 | 0.005 |
| Creatinine (mg/dL) | 0.74±0.21 | 0.74±0.16 | 0.74±0.24 | 0.688 |
| Uric acid (mg/dL) | 5.4±2.1 | 5.2±2.6 | 5.5±1.8 | 0.109 |
| Apolipoprotein A1 (mg/dL) | 0.43±0.26 | 0.42±0.22 | 0.44±0.27 | 0.544 |
| Apolipoprotein B (mg/dL) | 0.65±0.28 | 0.61±0.28 | 0.67±0.25 | 0.037 |
| C-reactive protein (mg/dL) | 6.54±17.7 | 4.80±13.7 | 7.5±19.5 | 0.064 |
| Fibrinogen (g/L) | 291.9±144.5 | 297±134.1 | 289±150.1 | 0551 |
| Lipoprotein (a) (mg/dL) | 39.0±41.2 | 31.4±32.1 | 43.3±45.0 | <0.001 |
| Protein-S % | 72.1±22.0 | 63.4±23.0 | 73.0±21.7 | 0.038 |
| Protein-C % | 66.3±26.6 | 63.2±29.6 | 66.6±26.2 | 0.537 |
| Antithrombin III % | 80.1±18.5 | 80.4±26.4 | 80.1±17.8 | 0.966 |
| Factor V activity % | 49.9±37.5 | 49.7±38.3 | 49.9±37.0 | 0.953 |
Clinical and biochemical data of all subjects according to the genotype of the LTA 252A>G polymorphism.
| LTA 252A>G | AA N = 247(47.1) | AG N = 225(42.9) | GG N = 52(10) | p-value |
|---|---|---|---|---|
| Age (years) | 40.4±5.3 | 40.8±5.4 | 40.1±5.1 | 0.274 |
| Women (%) | 120(48.6) | 127(56.4) | 24(44.2) | 0.126 |
| Hypertension | 58(23.5) | 74(32.9) | 17(32.7) | 0.060 |
| Smokers | 100(40.5) | 96(42.7) | 19(37.2) | 0.749 |
| Diabetes | 37(15.1) | 39(17.3) | 11(21.1) | 0.533 |
| Dyslipidemia | 135(54.7) | 131(58.5) | 32(61.5) | 0.551 |
| Family history of CAD | 116(47.3) | 107(47.8) | 32(61.5) | 0.159 |
| Coronary artery disease | 143(57.9) | 154(68.4) | 38(73.1) | 0.021 |
| Body mass index (kg/m2) | 25.8±6.2 | 25.4±5.7 | 24.0±4.4 | 0.181 |
| Leukocytes (mm3) | 6723±1711 | 6524±1767 | 6863±1811 | 0.329 |
| Platelets (mm3) | 186,313±118,890 | 176,638±103,955 | 182,634±114,614 | 0.646 |
| Cholesterol (mg/dL) | 114.9±66.7 | 116.6±61.7 | 115.6±63.6 | 0.960 |
| HDL-cholesterol (mg/dL) | 43.7±13.3 | 40.8±12.5* | 40.2±15.8 | 0.038 |
| LDL-cholesterol (mg/dL) | 67.2±34.3 | 72.3±34.8 | 66.7±33.4 | 0.379 |
| Triglycerides (mg/dL) | 137.0±289.5 | 113.5±108.0 | 150.1±151.2 | 0.446 |
| Glucose (mg/dL) | 96.0±32.2 | 95.7±30.4 | 98.4±40.6 | 0.913 |
| Creatinine (mg/dL) | 0.76±0.18 | 0.72±0.24* | 0.73±0.20 | 0.133 |
| Uric acid (mg/dL) | 5.5±2.3 | 5.4±1.9 | 5.1±1.9 | 0.519 |
| Apolipoprotein A1 (mg/dL) | 0.43±0.25 | 0.44±0.26 | 0.42±0.27 | 0.793 |
| Apolipoprotein B (mg/dL) | 0.66±0.27 | 0.63±0.26 | 0.64±0.23 | 0.575 |
| C-reactive protein (mg/dL) | 5.5±15.0 | 5.8±12.4 | 12.0±32.0** | 0.030 |
| Fibrinogen (g/L) | 292.2±143.4 | 283.6±145.2 | 323.1±144.8 | 0.211 |
| Lipoprotein(a) (mg/dL) | 40.2±39.8 | 37.8±39.8 | 38.9±52.9 | 0.816 |
| Protein-S % | 70.8±21.7 | 73.3±21.5 | 72.2±24.9 | 0.683 |
| Protein-C % | 66.8±28.5 | 67.8±24.4 | 56.9±26.5 | 0.172 |
| Antithrombin III % | 79.8±20.0 | 81.3±16.5 | 77.1±19.6 | 0.517 |
| Factor V activity % | 52.8±37.6 | 45.5±37.3* | 53.8±36.3 | 0.107 |
Values in parenthesis are percentages. *HDL (AA vs. AG), p = 0.020; creatinine (AA vs. AG), p = 0.046; FV (AA vs. AG), p = 0.048; ** C-reactive protein (AA vs. GG), p = 0.010, and (AG vs. GG), p = 0.014.
Females, smokers, and individuals with hypertension, diabetes, dyslipidemia, and a family history of CAD were significantly more prevalent in the CAD group. Compared with the controls, the CAD patients exhibited significantly lower plasma concentrations of HDL-cholesterol (p<0.001) and higher levels of fasting glucose (p = 0.005), lipoprotein (a) (p<0.001), and protein S (p = 0.038). The genotypic frequency distributions of the LTA A252G polymorphism for A/A homozygotes, A/G heterozygotes, and G/G homozygotes were 47.1%, 42.9, and 10% for all subjects; 55.0%, 37.6%, and 7.4% for the control group; and 42.7%, 46.0%, and 11.3% for the patient group, respectively (p = 0.021). The frequencies of the A and G alleles of the LTA gene were 68.6% and 31.4% among the 524 individuals in the entire population; 73.8% and 26.2% in the controls; and 65.7% and 34.3% in the CAD group, respectively. Genotyping distributions in both the control and CAD groups were consistent with Hardy-Weinberg equilibrium.
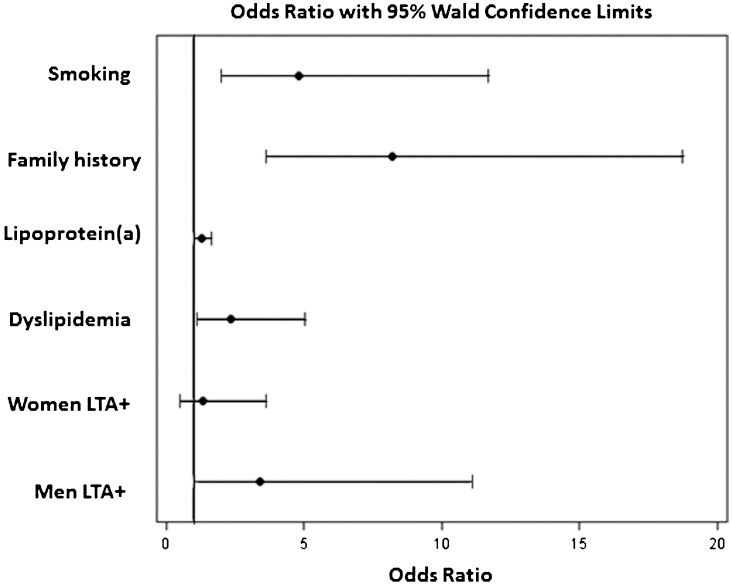
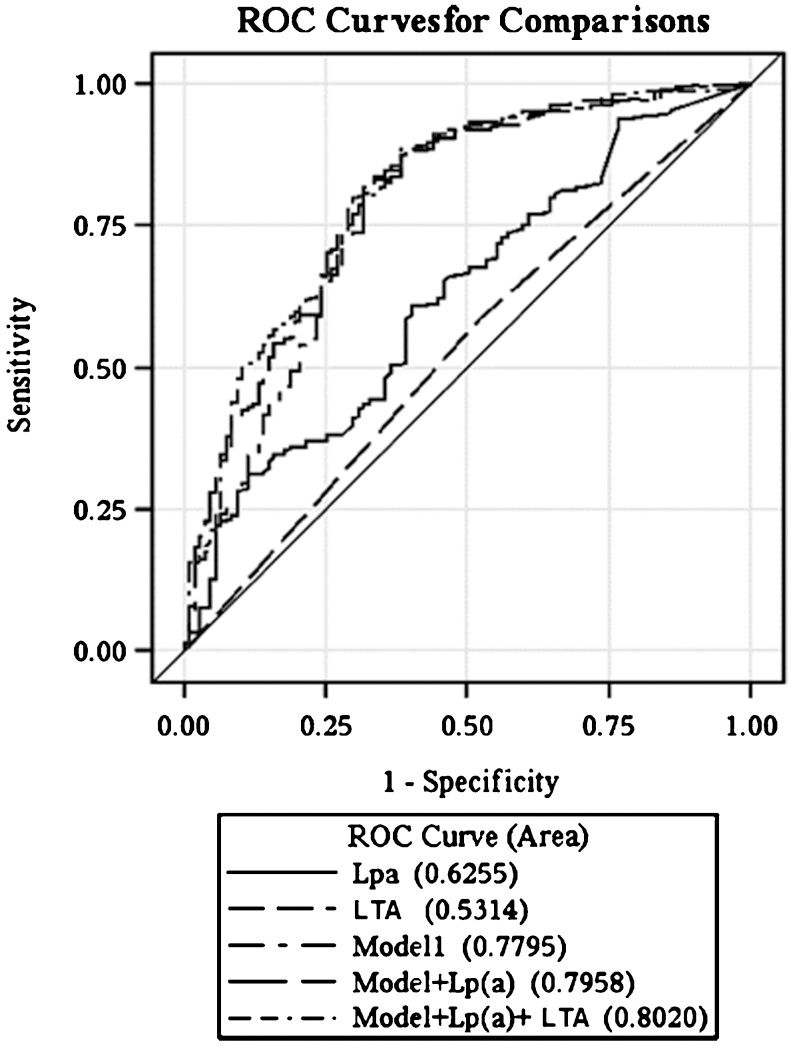
No association was observed between LTA A252G and traditional risk factors. HDL-cholesterol and C-reactive protein plasma levels were higher in the AA (p =0.038) and GG genotype subjects (p =0.030), respectively. A multivariate logistic regression analysis showed that smoking (OR 4.81; 95% CI 1.98 – 11.7, p =0.001), dyslipidemia (OR 2.35; 95% CI 1.10 – 5.05, p =0.028), family history of CAD (OR 8.20; 95% CI 4.692 - 12.103, p<0.001), and Lp(a) (OR 1.29; 95% CI 1.01 - 1.65, p =0.042) and LTA (OR 3.38; 95% CI 1.03 – 11.10, p =0.007) mutations in men, but not in women (OR 1.34; 95% CI 0.49 – 3.63, p =0.567), were independent factors associated with CAD (Figure 1). The performances of the predictive risk models are shown in the ROC curves (Figure 2). Model1 included the clinical variables gender, hypertension, smoking, diabetes, family history of CAD, dyslipidemia, and body mass index. The AUC (C-statistic) of this model for the prediction of CAD in patients <50 years old was 0.779. There was a progressive increase in the AUC with the inclusion of the Lp(a) and Lp(a) + LTA mutation variables of 0.796 (p = 0.047, for comparisons between model1 and model1+Lp(a)) and 0.802 (p = 0.024, for comparisons between model1 and model1+Lp(a)+LTA mutation).

 curves. Model1 included the following variables: gender, hypertension, smoking, diabetes, family history of CAD, dyslipidemia, and body mass index. Model1 vs. Model1+Lp(a) (p = 0.047); Model1 vs. Model1+Lp(a)+LTA (p = 0.024).")
Our study shows that Lp(a) and LTA mutations in men are associated with premature CAD. Lp(a) and LTA mutations exhibit an additive effect on the C-statistic AUC for the traditional risk factors. Several studies have shown conflicting results regarding the association between specific biomarkers and the prediction of CAD (22-27).
Lipoprotein (a) and risk prediction for CADIn a nested case-control study, Kim et al. (28) analyzed 18 biomarkers previously associated with CAD in 321 patients with CAD and 743 control postmenopausal women. Five (vWF, factor VIII, homocysteine, IL-6, and D-dimer) of the 18 biomarkers tested were associated with CAD, but only D-dimer improved the C-statistic compared with traditional risk factors. Contrary to our results, Lp(a) was not associated with CAD, but the previous study included only women who were older (aged between 50 and 79 years) than our patients. Blakenberg et al. (29) analyzed the risk prediction of CAD associated with 30 biomarkers in 2 middle-aged European populations. The study analyzed Lp(a) and other biomarkers and showed that no biomarker improved risk estimation in either population. The structure of lipoprotein (a) is similar to the structure of LDL cholesterol, which includes one additional plasminogen-like glycoprotein, namely apolipoprotein (a). Due to these characteristics, this particle may contribute to the processes of atherogenesis and thrombogenesis. Lp(a) studies showed conflicting results regarding the risk prediction of CAD (30-34). Bennet et al. (31) showed that the odds ratios for CAD were progressively higher with increasing Lp(a) levels in a large, prospective, population-based cohort that was adjusted for several established risk factors. Virani et al. (33) also showed that Lp(a) levels are associated with an increased risk of cardiovascular diseases in both African American and Caucasian male and female subjects. Sawage et al. (34) demonstrated an association between low Lp(a) levels and all-cause or cancer deaths and similar outcomes for low and high Lp(a) levels for cardiovascular deaths.
Use of genetic markers in risk prediction for CADFew studies have utilized genetic markers of CAD in risk prediction. Rossouw et al. (35) analyzed 23 inflammatory, lipid, and thrombotic biomarkers and 8 genetic polymorphisms in postmenopausal women (aged 50-79 years) receiving hormone replacement therapy in a nested case-control study. Of the 23 biomarkers, the following 13 were associated with CHD events: interleukin 6, matrix metalloproteinase 9, HDL-C, LDL-C, total cholesterol, triglycerides, D-dimer, factor VIII, von Willebrand factor, leukocyte count, homocysteine, fasting insulin, and 1 genetic polymorphism (the glycoprotein IIIa leu33pro mutation). Additive risk prediction of the biomarkers over the conventional risk factors was not performed. Ripatti et al. (36) calculated a genetic risk score based on 13 SNPs and observed an association with a first coronary heart disease event, but the genetic risk score did not improve risk prediction compared with risk prediction based on traditional risk factors and family history. Paynter et al. (37) calculated a genetic risk score based on 101 SNPs in 19,313 healthy Caucasian women who were followed up over a median of 12.3 years in a prospective study. After adjustment for traditional cardiovascular risk factors, the genetic risk score was not associated with cardiovascular disease risk. The LTA gene mutation was not included in the genetic risk score of the two previous studies. Thanassoulis et al. (38) showed a marginal increase in the risk prediction of cardiovascular events with the genetic risk score in addition to standard cardiovascular risk factors and a high risk of coronary artery calcium. However, future studies evaluating the utility of coronary artery calcium and genetic risk scores in predicting lifetime risk are needed. Genome-wide single nucleotide polymorphism association studies have identified several SNPs that are significantly associated with CAD and with traditional risk factors for CAD (39-42). However, the results of all these studies still showed inconsistencies with regard to the presence of a causal relationship between biomarkers, with or without the inclusion of genetic markers, and CAD and traditional risk factors. A mechanistic basis for the association between SNPs and coronary heart disease was observed, although the atherosclerotic process was unknown (43). However, none of the previously mentioned studies analyzed multiple biomarkers for the prediction of CAD in young adults aged <50 years.
In this study, we analyzed several biomarkers related to thrombogenic and atherogenic processes, and we included the LTA mutation as a candidate gene approach to risk assessment. The LTA mutation was frequently associated with CAD. In our study, the presence of the LTA gene mutant allele provided incremental information about CAD risk prediction. Previous case-control and cross-sectional studies examined the association between LTA gene polymorphisms and cardiovascular disease, but the results were inconsistent. The study by Ozaki et al. (17) described significant associations between LTA gene polymorphisms and myocardial infarction; however, the authors did not adjust for relevant covariates, including gender and age, and the genotype distributions among the control subjects were not in Hardy-Weinberg equilibrium. The association between LTA gene polymorphisms and CHD was confirmed in another Japanese population and in the family-based European PROCARDIS (precocious coronary artery disease) study (44). A significant association was observed between the LTA C804A genotype and the extent of coronary atherosclerosis in Caucasian patients with angiographically confirmed coronary atherosclerosis (45). However, several other studies did not detect an association between LTA gene polymorphisms and myocardial infarction (10,14,15). A meta-analysis performed by Clarke et al. (46) also showed no relationship between LTA gene polymorphisms and CHD. However, they did not study the LTA gene polymorphism (rs909253) evaluated in the Japanese study and our study. The most straightforward explanation for the findings of our study may be associated with the genetic differences of studied populations. Among populations with similar genetic backgrounds, the differences in allele frequency make it difficult to extrapolate genetic findings from one population to another. In our population, we demonstrated an association between LTA gene polymorphisms and increased risk of well-documented stable CAD in younger patients aged <50 years. Genetic diversity in different populations may be especially relevant to the LTA genomic region, which lies within the HLA region and has an erratic pattern of LD structure, with both short LD islands and long-range haplotypes (47). In our study, Lp(a) and LTA mutations alone were associated with lower risk prediction for CAD compared with conventional risk factors, but the inclusion of each mutation with conventional risk factors resulted in a progressive and statistically significant increase in risk prediction for CAD (Figure 2). The impact of the increased risk prediction based on AUC/C-statistics from 0.779 to 0.802 in clinical practice is uncertain and needs to be confirmed in future studies.
Study limitationsOur study has several limitations. This was a case-control study, and in such studies, casual associations are frequent when the sample size is relatively small for the association analysis of complex diseases with genetic mutations of multifactorial traits. Additionally, this case-control study was age-matched but not gender-matched. Therefore, the clinical and laboratory features observed more often in women may have influenced the final results. The analysis of one mutation can lead to results not identified in prospective studies with a larger number of individuals. The analysis of a significantly higher number of mutations, use of better laboratory techniques, and application of more sophisticated statistical models improved the genetic risk prediction for CAD. This study examined a significant number of atherogenic and thrombogenic markers in patients with premature CAD, but the markers represent a small portion of those involved in the atherosclerosis process. The complex interaction between genetic factors and the environment, including the impact of current medications on the treatment of CAD, may have further influenced the data interpretation.
Traditional risk factors played a strong role in risk prediction for the onset of premature coronary atherosclerosis in our population. The inclusion of Lp(a) and LTA mutations in the set of conventional risk factors showed an additive but small increase in the risk prediction for premature CAD.
ACKNOWLEDGMENTSFinancial support was provided by “Fundação de Amparo à Pesquisa do Estado de São Paulo (FAPESP)” number 01/06632-1.
Mansur AP contributed to the study conception and design, data analysis and interpretation, and writing of the manuscript. Takada JY performed the collection, analysis, and interpretation of data. Strunz CM performed the lab analysis and interpretation of data. Avakian SD contributed to the study design, as well as collection, analysis, and interpretation of data. César LA performed the analysis and interpretation of data. Ramires JA contributed to the study conception and design, as well as revision and final approval of the manuscript to be published.
No potential conflict of interest was reported.