








The clinical significance of ischemia/reperfusion of the lower extremities demands further investigation to enable the development of more effective therapeutic alternatives. This study investigated the changes in the vascular reactivity of the rabbit femoral artery and nitric oxide metabolites under partial ischemia/reperfusion conditions following cilostazol administration.
METHODS:Ischemia was induced using infrarenal aortic clamping. The animals were randomly divided into seven groups: Control 90 minutes, Ischemia/Reperfusion 90/60 minutes, Control 120 minutes, Ischemia/Reperfusion 120/90 minutes, Cilostazol, Cilostazol before Ischemia/Reperfusion 120/90 minutes, and Ischemia 120 minutes/Cilostazol/Reperfusion 90 minutes. Dose-response curves for sodium nitroprusside, acetylcholine, and the calcium ionophore A23187 were obtained in isolated femoral arteries. The levels of nitrites and nitrates in the plasma and skeletal muscle were determined using chemiluminescence.
RESULTS:Acetylcholine- and A23187-induced relaxation was reduced in the Ischemia/Reperfusion 120/90 group, and treatment with cilostazol partially prevented this ischemia/reperfusion-induced endothelium impairment. Only cilostazol treatment increased plasma levels of nitrites and nitrates. An elevation in the levels of nitrites and nitrates was observed in muscle tissues in the Ischemia/Reperfusion 120/90, Cilostazol/Ischemia/Reperfusion, and Ischemia/Cilostazol/Reperfusion groups.
CONCLUSION:Hind limb ischemia/reperfusion yielded an impaired endothelium-dependent relaxation of the femoral artery. Furthermore, cilostazol administration prior to ischemia exerted a protective effect on endothelium-dependent vascular reactivity under ischemia/reperfusion conditions.
The mechanisms of arterial occlusion are clearly associated with endothelial function, and these mechanisms are strongly affected by ischemia and blood reperfusion. Endothelial dysfunction is caused by impaired nitric oxide (NO) release or a decrease in NO bioavailability, which increases the risk of blood vessel spasm and thrombosis.
Cilostazol exerts a protective effect on various vascular tissues following acute ischemia (–3), and it alleviates the symptoms of intermittent claudication in peripheral vascular disease patients. This compound is a selective inhibitor of phosphodiesterase 3 (PDE3), which raises intracellular cyclic adenosine monophosphate (cAMP) content and activates protein kinase A (PKA). These events culminate in antiplatelet aggregation and peripheral vasodilation (4).
The role of the NO pathway on the pharmacological action of cilostazol remains largely unknown. Therefore, this study investigated the effects on NO/endothelium-dependent femoral reactivity in vitro, as induced by an in vivo cilostazol infusion. Rabbit hind limb global ischemia was induced using infrarenal abdominal aortic cross-clamping followed by reperfusion. The femoral artery wall response was examined because of its importance in the propagation of arterial blood throughout the hind limb.
METHODSDrugsAcetylcholine chloride (ACh), the calcium ionophore (A23187), sodium nitroprusside (SNP), phenylephrine (Phe), indomethacin, and cilostazol (Cil) were purchased from Sigma Chemical Company (St. Louis, MO, USA). SNP, ACh, and Phe were dissolved in distilled water. Indomethacin was dissolved in ethanol (10-5 M), and the calcium ionophore was first dissolved in dimethylsulfoxide (DMSO) and was subsequently diluted in water. Cilostazol was first dissolved in DMSO (10 mg/ml), and the 1 mg/kg dose was prepared by the dilution of this solution with saline to a volume of 5 ml. This dose was intravenously administered for 20 minutes (5).
Experimental designMale rabbits (2 2.5 kg) were pre-anesthetized with ketamine hydrochloride (Ketalar®, 5 mg/kg IM) and xylazine hydrochloride (Rompun®, 10 mg/kg IM), and they were anesthetized using pentobarbital sodium (30 mg/kg IV). The animals underwent tracheostomy, and they were ventilated using an endotracheal tube (3.0 mm, Rusch, Teleflex Medical, Durham, NC, USA) with 100% O2 in a pressure-controlled mode (Takaoka 600, K. Takaoka Indústria e Comércio Ltda, São Bernardo do Campo, SP, Brazil). The ear marginal artery was cannulated for volemic reposition with saline solution (NaCl 0.9%, 10 ml/kg/h). The abdominal infrarenal aorta was exposed using a median transperitoneal laparotomy approach, and a vascular clamp was applied to produce ischemia. The Institutional Animal Care and Use Committee of the School of Medicine of Ribeirão Preto, University of São Paulo, Brazil reviewed and approved the procedures for animal handling, which were in accordance with the Guide for the Care and Use of Laboratory Animals published by the U.S. National Institutes of Health (NIH Publication No. 85-23, revised 1996).
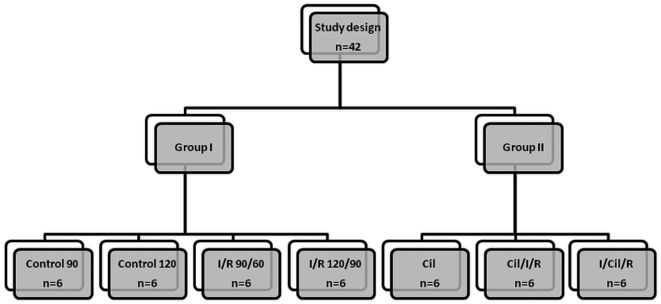
The animals were randomly divided into two main experimental groups, specifically, those treated without cilostazol (Group I - 4 subgroups) and with cilostazol (Group II - 3 subgroups): 1) Control 90 (anesthesia for 150 minutes); 2) Control 120 (anesthesia for 210 minutes); 3) I/R 90/60 (90 minutes of ischemia followed by 60 minutes of reperfusion); 4) I/R 120/90 (120 minutes of ischemia followed by 90 minutes of reperfusion); 5) Cil (endovenous cilostazol administered after anesthesia); 6) Cil/I/R (endovenous cilostazol administered before ischemia and submission of rabbits to 120 minutes of ischemia followed by 90 minutes of reperfusion); and 7) I/Cil/R (120 minutes of ischemia followed by cilostazol administration and reperfusion). Subgroups 1 and 2 from Group I were sham operation groups with different anesthesia times (120 and 210 minutes, respectively). Subgroups 3 and 4 from Group I (without cilostazol) underwent different durations of ischemia (90 and 120 minutes, respectively). Subgroups 5, 6, and 7 from Group II (with cilostazol) were used to investigate the isolated action of cilostazol and its protective effect when infused prior to or after 120 minutes of ischemia followed by 90 minutes of reperfusion (Figure 1).
 and Group II (with cilostazol): 1) Control 90 (anesthesia for 150 minutes); 2) Control 120 (anesthesia for 210 minutes); 3) I/R 90/60 (90 minutes of ischemia followed by 60 minutes of reperfusion); 4) I/R 120/90 (120 minutes of ischemia followed by 90 minutes of reperfusion); 5) Cil (endovenous cilostazol administered after anesthesia); 6) Cil/I/R (endovenous cilostazol administered before ischemia and submission of rabbits to 120 minutes of ischemia followed by 90 minutes of reperfusion); and 7) I/Cil/R (120 minutes of ischemia followed by cilostazol administration and reperfusion).")
Diagram of study design illustrating the following subgroups within Group I (without cilostazol) and Group II (with cilostazol): 1) Control 90 (anesthesia for 150 minutes); 2) Control 120 (anesthesia for 210 minutes); 3) I/R 90/60 (90 minutes of ischemia followed by 60 minutes of reperfusion); 4) I/R 120/90 (120 minutes of ischemia followed by 90 minutes of reperfusion); 5) Cil (endovenous cilostazol administered after anesthesia); 6) Cil/I/R (endovenous cilostazol administered before ischemia and submission of rabbits to 120 minutes of ischemia followed by 90 minutes of reperfusion); and 7) I/Cil/R (120 minutes of ischemia followed by cilostazol administration and reperfusion).
The femoral artery was carefully exposed through an anterior-medial hind limb incision and was immersed in Krebs solution composed of the following (in mM): NaCl 118.3, KCl 4.7, MgSO4 1.66, KH2PO4 1.22, CaCl2 2.5, NaHCO3 25.0, and glucose 11.1. Femoral artery segments (4-5 mm in length) were prepared with caution to avoid damage to the intimal surface of the vascular segments. The endothelium was removed by the gentle rubbing of the intimal surface of the blood vessel with a pair of watchmaker's forceps to investigate vascular smooth muscle function without the influence of the endothelium. This procedure removes the endothelium, but it does not affect vascular smooth muscle contraction. Vascular segments with and without endothelium were suspended in organ chambers (10 ml) filled with Krebs solution, maintained at 37°C and bubbled with 95% O2/5% CO2 (pH = 7.4). Each ring was suspended using two stainless steel clips that passed through the lumen. One clip was anchored to the bottom of the organ chamber, and the other clip was connected to a strain gauge for the measurement of isometric force (Grass FT03; Grass Instrument Company, Quincy, MA). Each ring was stretched to the optimal length-tension of 2 g (determined in a pilot study) and equilibrated for 60 minutes. Tissues were washed every 20 minutes during the equilibration period. Endothelial integrity was assessed by the degree of ACh-induced (10-6 M) relaxation in rings that were precontracted with Phe (10-6 M). The endothelium was present when the ACh-induced relaxation was 80% or greater. Each ring was washed and re-equilibrated for 30 minutes in the presence of indomethacin (10-5 M) to exclude the cyclooxygenase effects from the analysis because the NO pathway was the target of the investigation. The femoral artery rings were preincubated with Phe (10-6 M), and dose-response curves for SNP, ACh, and A23187 were obtained after the rings reached a stable plateau.
Nitrite and nitrate (NOx) quantificationBlood samples from the cava vein were collected in tubes containing heparin (1∶20 v/v). The blood was centrifuged (3000×g, 10 minutes, 4°C), and plasma aliquots were stored at -70°C for subsequent NOx measurement. Muscle (gracilis) tissue samples were wrapped and immediately stored at -70°C. The muscle samples were homogenized in Tris-HCl (20 mM, pH 7.4, 10% w/v) for analysis. The homogenate was centrifuged (3000×g, 10 minutes, 4°C), and the supernatant was used to measure NOx concentrations and total protein using the modified biuret reaction (6). Plasma and muscle samples were analyzed using an ozone-based chemiluminescence assay. Briefly, the samples were treated with cold ethanol (1∶2 v/v for 30 minutes at -20°C) and centrifuged (4000×g, 10 min). NOx levels were measured by injecting 25 µL of the supernatant into a glass purge vessel containing 0.8% vanadium (III) in HCl (1 N) at 90°C, which reduces NOx to an NO gas. A nitrogen stream was bubbled through the purge vessel, which contained vanadium (III), and through NaOH (1 N) into an NO analyzer (Sievers® Nitric Oxide Analyzer 280, GE Analytical Instruments, Boulder, CO, USA). The NOx concentration was calculated from a standard curve (sodium nitrate 0.5, 1.5, 10, and 50 mM), and it is expressed in µM for plasma and µM/mg protein for muscle samples.
Statistical analysisThe results are expressed as means ± the standard error of the mean (SEM). The dose-response curves for ACh, A23187, and SNP were derived using molar concentrations, and the logarithm of the molar concentration (log [M]) is presented in the three figures of figure 3. The EC50 was calculated from the dose-response curves of SNP), ACh, and A23187. The concentration-response curves were analyzed using two-way repeated-measures analysis of variance (ANOVA) and Bonferroni post-hoc tests. The concentrations of NOx were assessed using a one-way ANOVA (Prism 4.0, GraphPad Software, Inc., San Diego, CA, USA). Values were considered to be statistically significant at p<0.05.
RESULTSVascular reactivity ACh (10-10 to 3.10-5 M); (B) A23187 (10-10 to 3.10-5 M); and (C) SNP (10-10 to 3.10-5 M) from the Control and Cilostazol groups. * Indicates the difference between the Control 120 and Cil groups (p<0.05).")
ACh and A23187 promoted concentration-dependent relaxation in femoral artery endothelium-intact rings, and this relaxation was reduced in the I/R 120/90 group (Figures 2A and 2B). SNP produced a concentration-dependent relaxation in femoral artery endothelium-denuded rings, and no intergroup statistical differences were observed (Figure 2C). Cilostazol treatment impaired ACh- and A23187-induced relaxation (Figures 3A and 3B), but cilostazol did not alter SNP-induced relaxation (Figure 3C). Cilostazol administration prior to the induction of ischemia partially prevented the ischemia/reperfusion-induced impairment of ACh- and A23187-induced relaxation (Figures 4A and 4B). No change in SNP-induced relaxation was observed (Figure 4C). The EC50 values of sodium nitroprusside (SNP), acetylcholine (ACh), and the calcium ionophore (A23) are listed in Table 1.
 ACh (10-10 to 3.10-5 M); (B, E) A23187 (10-10 to 3.10-5 M); and (C, F) SNP (10-10 to 3.10-5 M) from the Control and I/R groups. * Indicates the difference between the Control 120 and I/R 120/90 groups (p<0.05).")
 ACh (10-10 to 3.10-5 M); (B, E) A23187 (10-10 to 3.10-5 M); and (C, F) SNP (10-10 to 3.10-5 M) from the Control, ischemia/reperfusion, and ischemia/reperfusion with cilostazol groups. * Indicates a difference from the Control 120 group, and # indicates a difference from the I/R 120/90 group (p<0.05).")
Femoral artery concentration-response curves. (A, D) ACh (10-10 to 3.10-5 M); (B, E) A23187 (10-10 to 3.10-5 M); and (C, F) SNP (10-10 to 3.10-5 M) from the Control, ischemia/reperfusion, and ischemia/reperfusion with cilostazol groups. * Indicates a difference from the Control 120 group, and # indicates a difference from the I/R 120/90 group (p<0.05).
The EC50 values of sodium nitroprusside (SNP), acetylcholine (ACh), and the calcium ionophore (A23). The data are expressed as means±SEM.
| Cont 90 | I/R 90 | Cont 120 | I/R 120 | Cil | Cil/I/R | I/Cil/R | |
|---|---|---|---|---|---|---|---|
| ACh | 6.28±0.03 | 6.23±0.04 | 6.31±0.03 | 5.98±0.03 | 6.14±0.03 | 6.47±0.03 | 5.99±0.03 |
| SNP | 5.96±0.01 | 5.95±0.01 | 5.93±0.02 | 5.96±0.03 | 5.69±0.08 | 5.88±0.02 | 5.92±0.02 |
| A23 | 6.43±0.01 | 6.40±0.01 | 6.43±0.01 | 6.52±0.04 | 5.98±0.14 | 6.20±0.06 | 6.57±0.03 |
No differences in the plasma levels of NOx (the Control 90 group versus the I/R 90/60 group and the Control 120 group versus the I/R 120/90 group) (Figure 5A) between the Control and I/R groups were observed. Cilostazol administration prior to and after ischemia increased plasma levels of NOx compared to levels in the Control 120 group (Figure 5B). In muscle tissues, 120 minutes of ischemia followed by 90 minutes of reperfusion increased the levels of NOx (Figure 6A). Cilostazol treatment prior to and after ischemia produced a stronger increased in NOx levels compared to the levels found in the Control 120 and I/R 120/90 subgroups (Figure 6B).
 and Control, ischemia/reperfusion and ischemia/reperfusion with cilostazol (B) groups. * Indicates a difference from the Control 120 group (p<0.05).")
 and Control, ischemia/reperfusion and ischemia/reperfusion with cilostazol (B) groups. * Indicates a difference from the Control 120 group (p<0.05).")
The re-establishment of blood flow to ischemic tissues or organs (i.e., reperfusion) is an essential step in many surgical procedures. However, reperfusion often leads to I/R injury, especially after a long period of ischemia (7). I/R injury is characterized by an increase in free reactive oxygen metabolites and cytokines, impaired NO production, the activation of adhesion molecules and neutrophils, and the promotion of lipid peroxidation with endothelial function impairment (8). The present study confirmed the importance of I/R duration on endothelium dysfunction; namely, an impairment of endothelium relaxation was observed in the I/R 120/90 group but not in the I/R 90/60 group. This result agrees with our previous experimental investigations that demonstrated femoral artery endothelium dysfunction in an I/R 120/90 group using an infrarenal clamp but indicated no alteration in the endothelium-dependent relaxation of the celiac trunk, renal, and superior mesenteric arteries after I/R 60/30 of the supraceliac aorta (4,9).
The present study also demonstrated no significant differences in the plasma levels of NOx between the groups. An increase in the muscle levels of NOx was observed in the I/R 120/90 group, but no significant differences between the I/R and Cil groups were observed. The Cil/I/R group exhibited greater relaxation compared to the I/R group. The plasma levels of NOx were augmented in the Cil/I/R group.
The vascular reactivity results revealed that 120 minutes of ischemia induced by infrarenal aortic cross-clamping followed by 90 minutes of reperfusion reduced ACh- and A23187-induced endothelium-dependent relaxation, but these conditions did not affect the endothelium-independent relaxation of rabbit femoral arteries. These data are consistent with the results from two previous investigations that described relaxation dysfunction in canine femoral arteries after a 3 h/2 h hind limb I/R (4,10). Cilostazol administration prior to ischemia partially prevented the impaired ischemia/reperfusion-induced relaxation found in the current model. This endothelial dysfunction indicated that the loss of NO-mediated vasorelaxation is an initial manifestation of free radical release during reperfusion. Cilostazol treatment reduces the size of myocardial infarcts (3,11). Therefore, our results showing the impairment of an endothelium-dependent vasodilation suggest that the vascular dysfunction found in our model is related to a reduced production of NO.
The reperfusion of ischemic tissue induces an acute inflammatory response that results in necrosis and irreversible vascular endothelial cell injury. However, the precise role of the NO that is generated by iNOS and/or eNOS during ischemia and reperfusion is controversial. The pharmacological inhibition of eNOS NO synthesis has deleterious effects on myocardial ischemia and reperfusion injury, but NO donor treatment or the superinduction of iNOS attenuates reperfusion-induced myocardial infarction and improves contractile function (12–14). Also, iNOS inhibition abolishes the cardioprotection resulting from ischemic preconditioning (15–18). However, the enhanced release of NO may contribute to ischemia and reperfusion injury, and NO inhibition may exert cardioprotective effects (19–21).
Our data revealed that 120 minutes of ischemia followed by 90 minutes of reperfusion increased NOx levels in muscle tissues. However, this phenomenon was not observed in plasma samples. Forty-five minutes of rat kidney ischemia followed by 6 hours of reperfusion boosts plasma levels of nitrite and nitrate, which are reduced by an iNOS inhibitor (22). Wild-type mice exhibit increased plasma NOx levels after myocardial I/R, but NOx species are not detected in iNOS-deficient (iNOS−/−) mice (23.) In contrast, a decrease in NO below basal levels after hind limb I/R (2.5 hours/2 hours) has been observed (24). These results suggest that iNOS activation mediates the rise in NOx after reperfusion.
Cilostazol administration prior to or after ischemia attenuated vascular dysfunction and increased NOx levels in plasma and muscle tissues. Several agonists have been found to activate eNOS after an enhancement in intracellular free Ca2+ concentrations, but an alternative pathway for eNOS activation by the serine/threonine protein kinase, Akt, has also been demonstrated, which should lead to an augmented release of NO (11,25,26). Stimulation of the phosphatidylinositol-3-OH kinase/Akt pathway improves serine phosphorylation and induces eNOS activation and NO release.
Cilostazol activates Akt and eNOS to promote the protective effect of statins and reduce infarct size. In addition, cilostazol induces NO release in human aortic endothelial cells through PKA-dependent eNOS phosphorylation (26). Therefore, the cilostazol-induced production of NOx species in the present work may be mediated by eNOS phosphorylation by Akt, the activation of protein kinase A (PKA), due to increased intracellular cAMP or an unknown pathway.
Study limitationThe present study did not use a NOS blocker to evaluate the effect of cilostazol on NO bioavailability. However, a recent study investigated the effect of systemic cilostazol administration on the ischemia-induced neovascularization process in vivo and examined the potential involvement of endothelial nitric oxide synthase (eNOS) signaling in the cilostazol-regulated neovascularization of ischemic muscles. The NOS inhibitor l-NAME abrogated the involvement of eNOS signaling in the neovascularization process (27).
In conclusion, 120 minutes of infrarenal aortic clamping followed by 90 minutes of reperfusion culminates in endothelium dysfunction, as suggested by the impaired endothelium-dependent relaxation of the femoral artery. Moreover, cilostazol administration prior to ischemia exerted a protective effect on the endothelium-dependent vascular reactivity under ischemia/reperfusion conditions.
AUTHOR CONTRIBUTIONSSantos MRGA and Capellini VK contributed to the execution of the experiments and statistics. Celotto AC contributed to the execution of the experiments and to the article writing. Evora PRB and Piccinato CE contributed to the article writing. Joviliano EE was responsible for the project design and article writing.
We thank José Carlos Vanni and Agnes Afrodite Albuquerque Sumarelli for technical support. We are grateful to the Fundação de Amparo à Pesquisa do Estado de São Paulo (FAPESP) and the Fundação de Apoio ao Ensino, Pesquisa e Assistência do Hospital das Clínicas da Faculdade de Medicina de Ribeirão Preto da Universidade de São Paulo (FAEPA/HCFMRP/USP) for financial support.
No potential conflict of interest was reported.