



To evaluate the roles of oxidative stress and lipid peroxidation in the ventricular remodeling that is induced by tobacco smoke exposure after myocardial infarction.
METHODS:After induced myocardial infarction, rats were allocated into two groups: C (control, n=25) and ETS (exposed to tobacco smoke, n=24). After 6 months, survivors were submitted to echocardiogram and biochemical analyses.
RESULTS:Rats in the ETS group showed higher diastolic (C = 1.52 ± 0.4 mm2, ETS = 1.95 ± 0.4 mm2; p=0.032) and systolic (C = 1.03 ± 0.3, ETS = 1.36 ± 0.4 mm2/g; p=0.049) ventricular areas, adjusted for body weight. The fractional area change was smaller in the ETS group (C = 30.3 ± 10.1 %, ETS = 19.2 ± 11.1 %; p=0.024) and E/A ratios were higher in ETS animals (C = 2.3 ± 2.2, ETS = 5.1 ± 2.5; p=0.037). ETS was also associated with a higher water percentage in the lung (C = 4.8 (4.3–4.8), ETS = 5.5 (5.3–5.6); p=0.013) as well as higher cardiac levels of reduced glutathione (C = 20.7 ± 7.6 nmol/mg of protein, ETS = 40.7 ± 12.7 nmol/mg of protein; p=0.037) and oxidized glutathione (C = 0.3 ± 0.1 nmol/g of protein, ETS = 0.9 ± 0.3 nmol/g of protein; p=0.008). No differences were observed in lipid hydroperoxide levels (C = 0.4 ± 0.2 nmol/mg of tissue, ETS = 0.1 ± 0.1 nmol/mg of tissue; p=0.08).
CONCLUSION:In animals exposed to tobacco smoke, oxidative stress is associated with the intensification of ventricular remodeling after myocardial infarction.
Shortly after myocardial infarction (MI), enlargement of the left ventricle can occur as a result of infarct expansion, increasing the surface of the infarcted area by stretching and thinning the damaged region.1 This regional alteration in cavity size produces left ventricular chamber enlargement, which increases the wall stress on the remaining normal regions. This, in turn, stimulates the altered expression of proteins and myocyte hypertrophy. In addition, increased neurohormonal activation, for instance, activation of endothelin, angiotensin II, and phenylephrine, after MI may stimulate collagen synthesis, thus leading to myocardial fibrosis of non-infarcted areas that are remote from the infarction.1 This process of alteration in ventricular size, composition, and mass is known as ventricular remodeling.2,3
Given that the extent of remodeling is related to increased risk for progressive ventricular dysfunction and cardiovascular death after MI, reversal or prevention of further remodeling may be key to improving prognosis following MI. Therefore, several strategies have been used to attenuate the remodeling process.4–7 Furthermore, situations that intensify ventricular remodeling should be avoided in order to improve the follow up after MI.
In the rat model, previous nicotine administration was shown to increase the size of the left ventricular cavity following MI8. Likewise, in the same model, exposure to tobacco smoke resulted in a reduced maximum rate of pressure increase in the left ventricle.9 However, the mechanisms involved in this process are not yet known. It is accepted that the free radicals contained in cigarette smoke contribute to the adverse effects of smoking.10–12 In addition, there is mounting evidence that free radicals and oxidative stress play a pivotal role in the process underlying cardiac remodeling.13–14 Therefore, the objective of this study was to evaluate the role of select oxidative stress-related variables on the ventricular remodeling induced by tobacco smoke exposure after MI.
METHODSCoronary artery ligationAll experiments and procedures were performed in concordance with the National Institute of Health’s Guide for the Care and Use of Laboratory Animals and were approved by the Animal Ethics Committee of our institution.
Male Wistar rats weighing 200–250 g were used in the study. Myocardial infarction was induced as previously described15. In brief, under ether administration, the heart was exteriorized after left thoracotomy. The left atrium was retracted to facilitate ligation of the left coronary artery with 5–0 Mono Nylon between the pulmonary outflow tract and the left atrium. The heart was then placed in the thorax, the lungs were inflated by positive pressure, and the thoracotomy was closed. After surgery, the rats were housed in a temperature controlled room (24ºC) with a 12-hour light:dark cycle. Food and water were supplied ad libitum.
Groups and TreatmentTwo days after MI, the animals were allocated into two groups: Group C (n=25), infarcted rats that were not exposed to tobacco smoke, and Group ETS (n= 24), infarcted rats exposed to tobacco smoke (40 cigarettes/day).
The ETS rats were exposed to cigarette smoke in a chamber (dimensions 95x80x65 cm) connected to a smoking device constructed based on a model published by Wang et al.16 and adapted by Paiva et al.17 The smoke was drawn out of filtered commercial cigarettes (composition per unit: 1.1 mg nicotine, 14 mg tar, and 15 mg carbon monoxide) with a vacuum pump and exhausted into the smoking chamber. During the first week, the number of cigarettes was gradually increased from 5 to 10 cigarettes over a 30 min period, administered twice each afternoon. Subsequently, 10 cigarettes were used in each smoking trial, repeated four times/day, twice in the morning and twice in the afternoon.
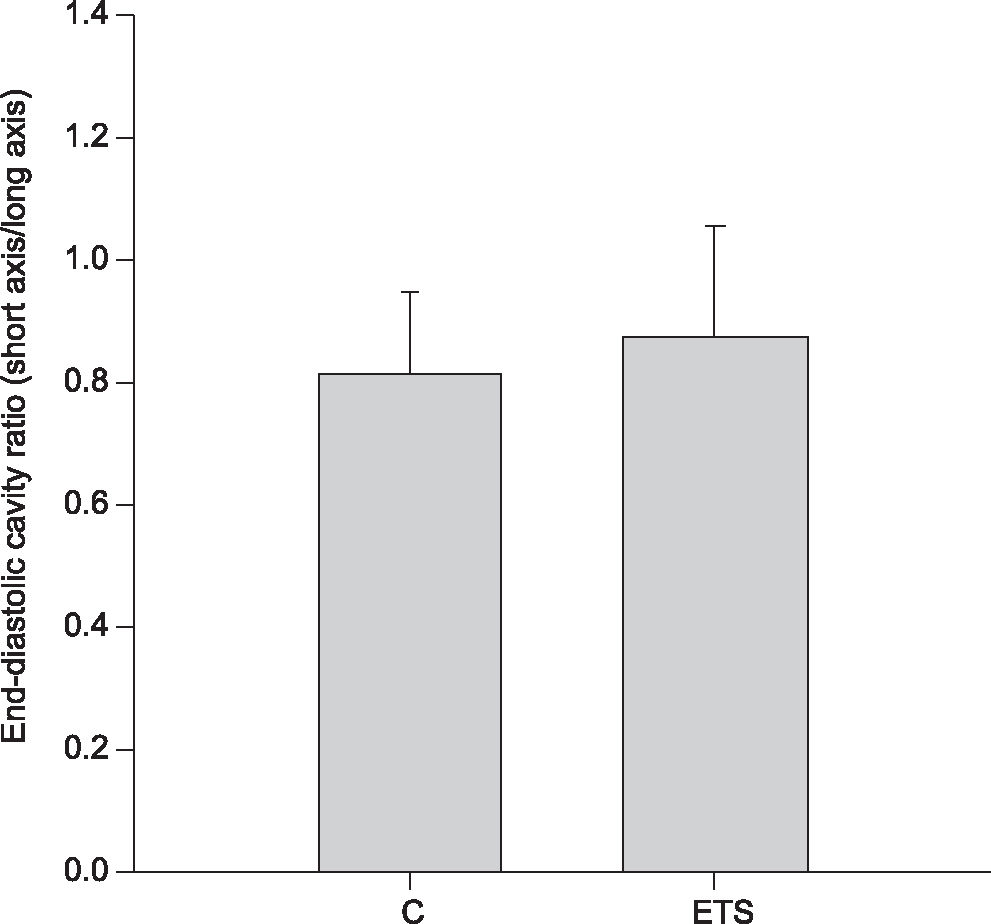
Echocardiographic StudyAfter 6 months of exposure, all animals were weighed and evaluated by a transthoracic echocardiograph exam. The exams were performed using a commercially available echocardiographic machine (Phillips TDI 5500) equipped with a 12 MHz phased array transducer. All measurements were made by the same observer according to the leading-edge method recommended by the American Society of Echocardiography.18 Measurements represented the mean of at least five consecutive cardiac cycles. LV end-diastolic dimension (LVEDD) and posterior wall thickness (LVWT) were measured at maximal diastolic dimension, and the end-systolic dimension (LVSD) was taken at the maximal anterior motion of the posterior wall. The left atrium was measured at its maximal diameter and the aorta at the end of diastole. Subsequently, end-systolic and end-diastolic endocardial borders were traced in both short-axis and long-axis views. The end-systolic (SA) and end-diastolic (DA) cavity areas were calculated as the sum of the areas from both the short and long-axis views in diastole (SumD) and systole (SumS), respectively. The ratio between the end-diastolic area in the short axis and the end-diastolic area in the long axis were also calculated. The fractional area change (FAC) was calculated from the composite cavity areas as: FAC = (SumD-SumS)/SumD.19 The velocities of transmitral diastolic flow (E and A velocities) were obtained in the apical four-chamber view. The E/A ratio was used as an index of LV diastolic function.
Morphometric analysisAt the completion of the functional study, the right and left ventricles (including the interventricular septum) were dissected, separated, and weighed.
The morphometric analysis of the myocardium was performed as described previously.20,21 Transverse 3 mm thick sections of LV were fixed in 10% buffered formalin and embedded in paraffin. Five μm thick sections were stained with hematoxylin-eosin and the collagen-specific stain picrosirius red (Sirius red F3BA in aqueous saturated picric acid). Myocyte cross-sectional area (CSA) was determined for at least 100 myocytes per slide stained with hematoxylin-eosin. The measurements were performed using a Leica microscope (400x magnification lens) attached to a video camera and connected to a personal computer equipped with image analyzer software (Image Pro Plus 3.0, Media Cybernetics, Silver Spring, MD). CSA was measured with a digitizing pad, and selected cells were cut transversely with the nucleus clearly identified in the center of the myocyte. The interstitial collagen volume fraction (IC) was determined for the entire picrosirius red-stained cardiac section using an automated image analyzer (Image Pro Plus 3.0, Media Cybernetics). Cardiac tissue components were identified according to the color level: red for collagen fibers, yellow for myocytes, and white for interstitial space. The digitized profiles were sent to a computer that calculated collagen volume fraction as the sum of all connective tissue areas divided by the sum of all connective tissue plus myocyte areas. On average, 35 microscopic fields were analyzed with a 40x lens. Both the infarcted area and perivascular collagen were excluded from this analysis. The lengths of the infarcted and viable muscle for both the endocardial and epicardial circumferences were determined by planimetry. Infarction size was calculated by dividing the endocardial and epicardial circumferences of the infarcted area by total epicardial and endocardial ventricular circumferences. Measurements were made in midventricular slices (5–6 mm from the apex) under the assumption that the left midventricular slice would show an approximately linear relationship to the sum of the area measurements from all heart slices.22
Oxidative stressTo analyze oxidative stress, we assessed the values of hepatic and cardiac reduced glutathione (GSH), oxidized glutathione (GSSG), GSH/GSSG ratio, and lipid hydroperoxide (LH), as described previously.23 The septum of the heart was subjected to the oxidative stress test. In brief, GSH was measured by a kinetic assay in reaction medium (0.6 mM DTNB, 0.2 mM NADPH and 2 U of glutathione reductase in 50 mM KH2PO4/5 mM EDTA). GSSG was assayed in the heart and liver in reaction medium (2 mM 5,5′-dithiobis-(2-nitrobenzoic) acid (DTNB) in 50 mM KH2PO4/5 mM EDTA). Lipid hydroperoxide was measured in the heart and liver through hydroperoxide-mediated oxidation of Fe2+ in a solution composed of 100 μl of sample and 900 μl of a reaction mixture (250 μM FeSO4, 25 mM H2SO4, 100 μM xylenol orange, and 4 mM butylated hydroxytoluene (BHT) in 90% (v/v) methanol).
Statistical analysisA Student’s t test was used to compare groups with normal distributions. In all other cases, comparisons between groups were made using the Mann-Whitney U test. Data are expressed as mean ± SD or medians (including the lower quartile and upper quartile). Survival data were analyzed with the Kaplan-Meyer curve and Cox regression model. Data analysis was carried out with SigmaStat for Windows v.2.03 (SPSS Inc., Chicago, IL). The level of significance was set at 5%.
RESULTSAfter the 6-month follow-up period, 12 control animals and 10 ETS animals had survived. This difference was not statistically significant (p>0.05).
The echocardiographic data are shown in Table 1 and Figure 1. Heart rate was significantly higher in ETS animals (C = 251 ± 29, ETS = 298 ± 39; p=0.006). The left atrial diameter, adjusted for body weight, was higher in ETS animals in comparison to controls (C = 12.03 ± 3.7 mm/kg, ETS = 14.96 ± 2.6 mm/kg; p=0.048). ETS animals showed higher diastolic (C = 1.52 ± 0.4 mm2, ETS = 1.95 ± 0.4 mm2; p=0.032) and systolic (C = 1.03 ± 0.3, ETS = 1.36 ± 0.4 mm2/g; p=0.049) ventricular areas, adjusted for body weight. The fractional area change was smaller in ETS animals (C = 30.3 ± 10.1%, ETS = 19.2 ± 11.1 %; p=0.024) and the E/A ratio was higher in ETS animals (C = 2.3 ± 2.2, ETS = 5.1 ± 2.5; p=0.037). Other echocardiographic variables and measurements of the geometry of the ventricular chamber were not statistically significantly different between the groups.
Echocardiographic variables measured in rats exposed to tobacco smoke and controls
| Variables | Control | ETS | P |
|---|---|---|---|
| HR | 251 ± 29 | 298 ± 39 | 0.006 |
| LA/BW (mm/g) | 12.03 ± 3.7 | 14.96 ± 2.6 | 0.048 |
| LVEDD/BW (mm/g) | 20.5 ± 3.6 | 23.9 ± 5.2 | 0.093 |
| LVWT (mm) | 1.5 ± 0.3 | 1.4 ± 0.3 | 0.356 |
| LVWT/ LVEDD | 0.14 ± 0.02 | 0.13 ± 0.03 | 0.339 |
| E (cm/s) | 73.8 ± 24.9 | 80.6 ± 13.3 | 0.450 |
| A (cm/s) | 47. 2 ± 26.3 | 21.7 ± 19.2 | 0.055 |
| E/A | 2.3 ± 2.2 | 5.1 ± 2.5 | 0.037 |
| DA/BW (mm2/g) | 1.52 ± 0.4 | 1.95 ± 0.4 | 0.032 |
| SA/BW (mm2/g) | 1.03 ± 0.3 | 1.36 ± 0.4 | 0.049 |
| FAC (%) | 30.3 ± 10.1 | 19.2 ± 11.1 | 0.024 |
ETS- exposed to tobacco smoke, HR: heart rate, LV: left ventricle, LA: left atrium, BW: body weight, LVEDD: LV end-diastolic dimension, LVWT: LV posterior wall thickness, FAC: fractional area change, E: peak velocity of early ventricular filling, A: peak velocity of transmitral flow during atrial contraction, DA: diastolic area, SA: systolic area. Data are expressed as mean ± SD.
, an index of chamber geometry. ETS: exposed to tobacco smoke. p> 0.05.")
Table 2 summarizes the morphological data. The ETS group displayed a higher right ventricular weight, adjusted for body weight (C = 0.7 ± 0.3 mg/kg, ETS = 1.4 ± 0.3 mg/ kg; p=0.004), and higher water percentage in the lung (C = 4.8 (4.3–4.8), ETS = 5.5 (5.3–5.6); p=0.013). There were no differences in the other variables (p>0.05).
Morphology of the heart and lung in rats exposed to tobacco smoke and controls
| Variables | Control | ETS | P |
|---|---|---|---|
| BW (g) | 542 ± 67 | 484 ± 77 | 0.073 |
| LVW/BW (mg/kg) | 2.7 ± 0.8 | 2.9 ± 0.5 | 0.573 |
| RVW/BW (mg/kg) | 0.7 ± 0.3 | 1.4± 0.3 | 0.004 |
| Lung w/d | 4.8 (4.3–4.8) | 5.5 (5.3–5.6) | 0.013 |
| CSA (μm2) | 240 (235–244) | 258 (245–259) | 0.076 |
| IC (%) | 4.06 ± 0.4 | 3.5 ± 0.8 | 0.257 |
| % MI | 45.9 ± 4.1 | 48.0 ± 5.1 | 0.311 |
ETS- exposed to tobacco smoke, LV: left ventricle, BW: body weight, LVW: left ventricular weight, RVW: right ventricular weight, w/d: wet/dry ratio, CSA: cross-sectional area, IC: interstitial collagen volume fraction. %MI: infarction size. Data are expressed as mean ± SD or as medians with the lower quartile and upper quartile.
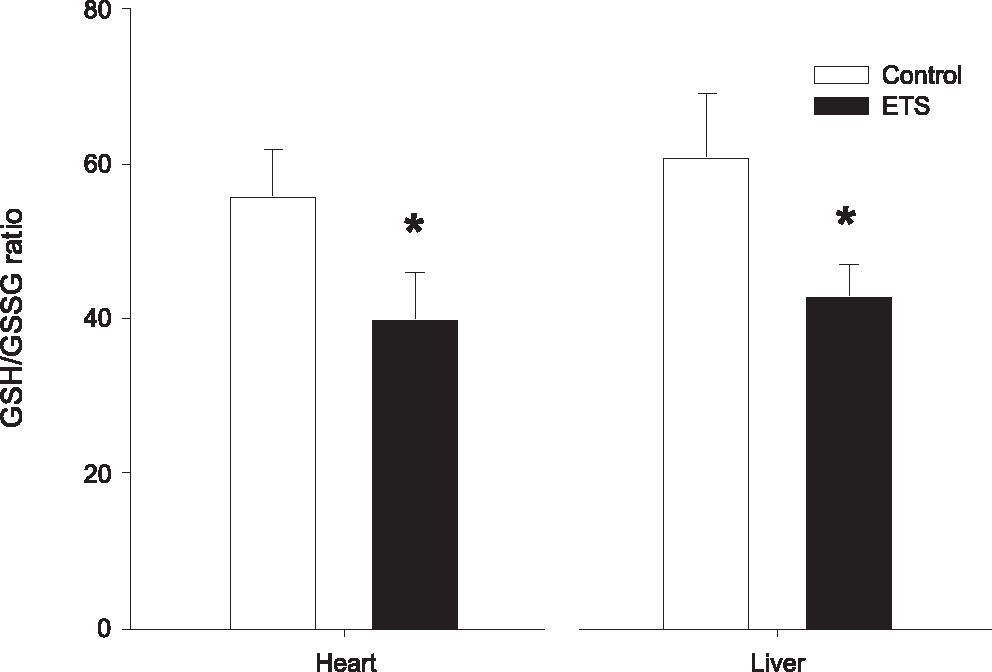
Table 3 summarizes the biochemical data. The ETS showed higher cardiac levels of reduced glutathione (C = 20.7 ± 7.6 nmol/g of protein, ETS = 40.7 ± 12.7 nmol/g of protein; p=0.037) and oxidized glutathione (C = 0.3 ± 0.1 nmol/g of protein, ETS = 0.9 ± 0.3 nmol/g of protein; p=0.008). No differences were observed in cardiac lipid hydroperoxide (C = 0.4 ± 0.2 nmol/mg of tissue, ETS = 0.1 ± 0.1 nmol/mg of tissue; p=0.08). Rats in the ETS group also showed higher levels of hepatic reduced glutathione (C = 17.7 ± 7.3 nmol/mg of protein, ETS = 33.7 ± 8.9 nmol/ mg of protein; p=0.033) and oxidized glutathione (C = 0.3 ± 0.1 nmol/g of protein, ETS = 0.8 ± 0.2 nmol/g of protein; p=0.009); no differences in hepatic lipid hydroperoxide were observed (C = 0.2 (0.2–0.4) nmol/mg of tissue, ETS = 0.2 (0.1–0.3) nmol/mg of tissue; p=0.20). The GSH/GSSG ratio was smaller in the ETS group (Figure 2) in both the heart (C = 56 ± 6, ETS = 40 ± 6; p=0.017) and the liver (C = 61 ± 8, ETS = 43 ± 4; p=0.010).
Measures of oxidative stress in rats exposed to tobacco smoke and controls
| Variables | Control | ETS | P |
|---|---|---|---|
| GSHc nmol/g of protein | 20.7 ± 7.6 | 40.7 ± 12.7 | 0.037 |
| GSSGc nmol/g of protein | 0.3 ± 0.1 | 0.9 ± 0.3 | 0.008 |
| LHc nmol/mg of tissue | 0.4 ± 0.2 | 0.1 ± 0.1 | 0.08 |
| GSHh nmol/g of protein | 17.7 ± 7.3 | 33.7 ± 8.9 | 0.033 |
| GSSGh nmol/g of protein | 0.3 ± 0.1 | 0.8 ± 0.2 | 0.009 |
| LHh nmol/mg of tissue | 0.2 (0.2–0.4) | 0.2 (0.1–0.3) | 0.20 |
ETS- exposed to tobacco smoke, c: cardiac, h: hepatic, GSH: reduced glutathione, GSSG: oxidized glutathione, LH: lipid hydroperoxide. Data are expressed as mean ± SD or medians with the lower quartile and upper quartile.

The objective of the present study was to analyze the contribution of oxidative stress on ventricular remodeling following myocardial infarction (MI) in rats exposed to tobacco smoke. Our results suggest that tobacco smoke exposure intensifies cardiac remodeling, and that this phenomenon may be modulated by oxidative stress.
The main finding of the present study was that ETS intensified the morphological changes in the left ventricle that are induced by coronary occlusion. This phenomenon was characterized mainly by an increase in left ventricular diameters compared with controls. Alterations in ventricular mass, volume, and geometry after cardiac injury can be interpreted as an expression of the remodeling process; regardless of the complexity of this process, measures of cardiac remodeling include heart size, shape and mass, and ventricular volumes.1–3 Therefore, our data indicate that ETS intensified the remodeling process following MI.
With regard to functional significance, ventricular remodeling is first and foremost a compensatory process.1–3 . However, chronic ventricular remodeling is now recognized as an important pathological process that causes progressive ventricular dysfunction and clinical presentation of heart failure or sudden death.1–3 Consistent with this model, our study showed that the intensification of the remodeling process in the ETS group resulted in decreased systolic cardiac function, as assessed by fractional area change. Likewise, the E/A ratio was higher in the ETS group, compatible with a restrictive pattern. Therefore, our results indicate that the intensification of the remodeling process in the ETS group was associated with impairment of both systolic and diastolic left ventricular function. As a consequence, the ETS rats presented signs of heart failure such as right ventricular hypertrophy and pulmonary congestion.
It is important to note that the mechanisms involved in the intensification of cardiac remodeling induced by ETS are not known. The present study rules out ventricular geometry as a pivotal modulator of cardiac remodelling induced by smoking. Likewise, although the extracellular matrix plays a critical role in the maintenance of ventricular geometry and function, we found that neither MMP-2 nor MMP-9 participate in ventricular remodeling induced by tobacco smoke exposure.24 Previous investigations did not show alterations in PO2 or in percentage of hemoglobin saturation upon exposure to tobacco smoke. Thus, chronic hypoxemia and alterations in blood viscosity secondary to hypoxia, which could explain our results, probably did not play a major role the pathophysiology of the cardiac alterations induced by cigarette smoke.25 Several studies provide strong evidence that exposure to tobacco smoke results in neurohormonal activation.26–28 In addition to its vascular effects, neurohormonal activation might, through autocrine and paracrine actions, activate intracellular signaling pathways and trigger cardiac hypertrophy. Finally, we should consider that a number of other factors might contribute to myocardial dysfunction of the remodeled heart, such as changes in contractile proteins, changes in the β-adrenergic pathway, abnormalities in calcium-handling proteins, increased apoptosis, metabolic remodeling, and alterations to gap junctions.29,30
Thus, we considered the potential role of oxidative stress on LV remodeling in response to tobacco smoke exposure after myocardial infarction. It is well recognized that an increase in free radical production and oxidative stress is induced by ETS. Conventionally, cigarette smoke is divided into two phases: a tar phase and a gas phase, the latter of which passes through the filter of a filtered cigarette. The tar phase contains >1017 free radicals/g, and the gas phase contains >1015 free radicals/puff. Another source of free radicals is the increased endogenous production induced by ETS. Irrespective of the mechanism, ETS is associated with the increased generation of free radicals.31 In addition, experimental studies have shown that antioxidant administration attenuates the cardiac alterations induced by ETS.33 Therefore, clinical and experimental observations suggest that oxidative stress induction is a potential mechanism involved in the etiology of smoking-related cardiovascular diseases.31,32
It is important to note that, in situations of oxidative stress, GSH is converted to GSSG. In this manner, decreased values of GSH and increased values of GSSG suggest oxidative stress. However, isolated GSH levels are not a reliable marker of oxidative stress, because GSH can be produced by a number of different systems, for instance, via glutathione synthase. Therefore, it is preferable to assay oxidative stress by a decreased GSH/ GSSG ratio and increased GSSG levels.33,34 Our study supports this assumption, since ETS was associated with a significant increase in GSSG, and a decreased GSH/ GSSG ratio. We also assessed liver glutathione because the liver plays a pivotal role in the response to oxidative stress through its important antioxidant defenses. In addition, the demonstration of redox alterations in both heart and liver suggests that the oxidative stress in this model is a systemic event.
It is well established that reactive oxygen species and oxidative stress play a critical role in cardiac remodeling following several cardiac injuries.13–14 Oxidative stress occurs when excess reactive oxygen species are generated (for example, due to activation of NADPH oxidase, mitochondrial electron transport, nitric oxide synthase, or xanthine oxidase) and cannot be adequately countered by antioxidant systems (such as the glutathione system, catalase, and superoxide dismutase). There are several mechanisms by which oxidative stress stimulates cardiac remodeling.35 For one, oxidative stress affects the extracellular matrix, activating metalloproteinases and stimulating cardiac fibroblast proliferation. Oxidative stress also activates a broad variety of transcriptional factors and remodeling kinases, and directly influences cardiac function by modifying contractile proteins. Importantly, oxidative stress triggers lipid peroxidation, which can lead to cell dysfunction and death.35 However, in our study, the lipid hydroperoxide levels measured in the ETS group were not significantly different from those in the control group. Therefore, although the exact mechanism involved in this process remains to be elucidated, our study suggests that lipid peroxidation does not contribute to the ETS-induced intensification of remodeling following MI. In conclusion, in animals exposed to tobacco smoke, oxidative stress is associated with ventricular remodeling intensification after acute myocardial infarction.
CONFLICT OF INTEREST: none declared