





It has been shown that SOCS-1 plays an important role in the proper control of cytokine/growth factor responses and acts as a tumor suppressor in acute myeloid leukemias. Therefore, the objective of the present study was to evaluate the in vitro effect of treatment with Nutlin-3, a small molecule inhibitor of the MDM2/p53 interaction, on the expression of the suppressor of cytokine signaling 1 in primary acute myeloid leukemia cells and in myeloid cell lines with differential p53 status.
METHOD:The expression of the suppressor of cytokine signaling 1 was quantitatively analyzed by real-time PCR in myeloid p53wild-type (OCI and MOLM) and p53null HL-60, leukemic cell lines, in patient-derived acute myeloid leukemia blasts, and in primary normal cell types, such as macrophages, endothelial cells, and bone marrow mesenchymal stem cells. The p53-dependence of the suppressor of cytokine signaling 1 upregulation that is induced by Nutlin-3 was analyzed in experiments performed using siRNA for p53, while the functional upregulation of the suppressor of cytokine signaling 1 was analyzed by assessing the levels of phosphorylated STAT-3.
RESULTS:Nutlin-3 significantly upregulated the transcription of the suppressor of cytokine signaling 1 in p53wild-type OCI and MOLM but not in p53deleted p53null HL60, myeloid leukemic cell lines, as well as in primary acute myeloid leukemia blasts. Conversely, and somewhat unexpectedly, Nutlin-3 did not modulate the suppressor of cytokine signaling 1 expression in primary normal macrophages, endothelial cells, and bone marrow mesenchymal stem cells. The p53-dependent upregulation of the suppressor of cytokine signaling 1 by Nutlin-3 was associated with the downregulation of phosphorylated STAT-3, a major molecular target of the suppressor of cytokine signaling 1.
CONCLUSION:Overall, our data suggest a potential role for the suppressor of cytokine signaling 1 as a therapeutic target of Nutlin-3 in p53 wild-type acute myeloid leukemias.
The selective small molecule inhibitor Nutlin-3 binds MDM2 in the p53 binding pocket with high selectivity and can release p53 from negative control, leading to an effective stabilization of p53 and activation of the p53 pathway (1). A number of studies have demonstrated that Nutlin-3 induces ex vivo cytotoxic cell death of p53wild-type acute myeloid leukemias (AMLs) (2–7), and Nutlin-3 was recently shown to upregulate the suppressor of cytokine signaling 1 (SOCS-1) in primary B leukemic cells through the mirR-155 pathway (8,9). Since the cloning of SOCS-1, it has become evident that the SOCS proteins are important for the proper control of cytokine and growth factor responses, and the absence of SOCS proteins leads to excessive cytokine signaling (10). In addition, SOCS-1 also acts as a tumor suppressor, as documented by the fact that SOCS-1 silencing by DNA hypermethylation at the gene promoter region has been found in both solid tumors, such as hepatocarcinomas (11), and AMLs (12–17).
Based on these findings, in the present study, we evaluated the effect of Nutlin-3 treatment on SOCS-1 expression in primary AML cells, as well as in myeloid cell lines with differential p53 status. Additionally, the effect of Nutlin-3 exposure on SOCS-1 expression was evaluated in primary normal cells characteristic of the bone marrow microenvironment, such as primary macrophages, endothelial cells, and multipotent stromal cells (MSCs).
MATERIALS AND METHODSCell cultureThe myeloid p53wild-type (OCI and MOLM) and p53null (HL-60) leukemic cell lines were purchased from the ATCC (American Type Culture Collection, Manassas, VA). MOLM and HL-60 leukemic cell lines were cultured in RPMI-1640 containing 10% FBS (both from Gibco BRL, Grand Island, NY), while OCI cells were cultured in alpha-MEM (LONZA, Basel, Switzerland) containing 10% FBS, as previously described (18).
Primary peripheral blood samples were collected in heparin-coated tubes from five AML patients and six healthy blood donors after informed consent was obtained in accordance with the Declaration of Helsinki and in agreement with institutional guidelines. Peripheral blood mononuclear cells (PBMC) from AML patients and healthy donors were isolated by gradient centrifugation with lymphocyte cell separation medium (Cedarlane Laboratories, Hornby, ON). The percentage of blasts among leukemic PBMC ranged from 60–85% for all patients, as assessed by light microscopy and verified by standard flow cytometry analysis. AML patient cells were seeded at a density of 1×106 cells/ml in RPMI containing 10% FBS (both from Gibco BRL). To obtain primary normal adherent macrophages, blood donor PBMCs were seeded at a density of 5×106 cells/ml, and non-adherent cells were removed after 18 hours. Adherent cells were cultivated in fresh RPMI medium containing 10% FBS, as previously described (19).
Human umbilical vein endothelial cells (HUVECs) were purchased from BioWhittaker (Walkersville, MD) and grown on 0.2% gelatin-coated tissue culture plates in M199 endothelial growth medium supplemented with 20% FBS, heparin, and 50 mg/ml ECGF (all from BioWhittaker), as previously described (20). In all experiments, cells were used between the 3rd and 5th passage in vitro. Bone marrow-derived MSCs were purchased from LONZA and grown in MSC Growth Medium (MSC-GM, LONZA), as previously described (21,22).
Culture treatments and evaluation of cell cytotoxicityCells were treated by adding Nutlin-3 (10 μM; obtained from Cayman Chemical, Ann Arbor, MI) to the culture medium. After treatment, cell viability was monitored up to 48 hours by Trypan blue dye exclusion, as previously described (23). In parallel, the degree of apoptosis was quantified by Annexin V-FITC/propidium iodide (PI) staining (Immunotech, Marseille, France) followed by flow cytometry analysis, as previously described (24,25). To analyze the cell cycle profile, cells were incubated with 50 μM 5-bromodeoxyuridine (BrdU; Sigma Aldrich) at 37°C for 1 hour, the anti-BrdU antibody (BD Biosciences Pharmingen, Franklin Lakes, NJ) was bound to BrdU, and the complex was detected by an FITC-conjugated secondary antibody (Beckman-Coulter, Marseille, France) (26,27). After additional staining with 50 μg/ml PI (Sigma-Aldrich), the cell samples were analyzed by flow cytometry.
RNA analysisAliquots of untreated and Nutlin-3-treated cells were harvested for RNA extraction 24–48 hours post-treatment. Total RNA was extracted using the Qiagen RNeasy Plus mini kit (Qiagen, Hilden, Germany) according to the supplier's instructions. The integrity of the total RNA preparation was assessed using an Agilent 2100 Bioanalyzer. RNA was transcribed into cDNA using the GEArray AmpoLabeling-LPR Kit (Superarray Bioscience Corporation, Frederick, MD). Investigations of SOCS-1 and miR-155 gene expression were both carried out in RNA samples with the real-time thermal analyzer Rotor-Gene™ 6000 (Corbett, Cambridge, UK) using SYBR Green-based technology and the RT-PCR primer set for human SOCS-1 cDNA or miR-155 (SABioscience, Frederick, MD). Gene expression of the target sequences was normalized with respect to the expression of endogenous controls. Each sample was tested in triplicate.
Western blot analysesCells were lysed in ice-cold RIPA buffer (50 mM Tris pH 7.5, 150 mM NaCl, 0.1% SDS, 1% Nonidet P-40, 0.25% sodium desoxycholate) supplemented with protease inhibitors (Complete, Roche; Germany) on ice for 1 hour (28). Before gel migration, samples were added to loading buffer (250 mM Tris pH 6.8, 2% SDS, 40% glycerin, 20% beta-mercaptoethanol) and boiled for 2 minutes. Equal amounts of protein for each sample were migrated in acrylamide gels and blotted onto nitrocellulose filters, as previously described (29), before incubation with the following monoclonal antibodies: anti-p53, anti-SOCS-1, anti-STAT-3, and anti-phospho-STAT-3 (all from Santa Cruz Biotechnology, Santa Cruz, CA), as well as anti-tubulin (Sigma-Aldrich). After incubation with peroxidase-conjugated anti-mouse IgG, specific reactions were revealed with the ECL detection kit (Amersham Pharmacia Biotech).
Multiplex immunoassayThe MILLIPLEX MAP Human Multi-Pathway 9-plex Magnetic Bead Signaling kit phosphoprotein (Merck Millipore, Billerica, MA, USA) was used to detect changes in phosphorylated ERK/MAP kinase 1/2, Akt, STAT-3, JNK, p70 S6 kinase, NF-kB, STAT-5A/B, CREB, and p38 in cell lysates using the Luminex system, according to the manufacturer's instructions. Briefly, cells were seeded at a density of 1×106/ml and treated with Nutlin-3 (10 μM). At different time points, cells were harvested in MILLIPLEX MAP lysis buffer (Merck Millipore) in the presence of the Protease Inhibitor Cocktail Set III (Calbiochem, San Diego, CA). Each lysate was diluted in the MILLIPLEX MAP Assay Buffer 2 (Merck Millipore), incubated at 4°C overnight, and analyzed according to the assay protocol. Median Fluorescence Intensity (MFI) was measured with the Luminex System and normalized for μg of protein.
Transfection experimentsOCI cells (1.25×106) were resuspended in 0.1 ml of Nucleofector™ solution V from the human Nucleofector kit V (Amaxa, Cologne, Germany). Two μg of plasmid DNA (GFP-construct) or 1 μg of siRNA was mixed with the 0.1 ml of cell suspension, transferred into a 2.0-mm electroporation cuvette, and nucleofected using an Amaxa Nucleofector II apparatus, following the manufacturer's guidelines and as previously described (30). After transfection, cells were immediately transferred into complete medium and cultured in six-well plates at 37°C. Transfection efficiency was estimated in each experiment by scoring the number of GFP-positive cells by flow cytometry analysis. siRNAs were designed and manufactured by Ambion Inc. (Austin, TX) according to the current guidelines for effective gene knock-down by this method and were validated in preliminary experiments. Negative control siRNA, comprised of a 19-bp scrambled sequence with 3′ dT overhangs (Ambion's Silencer negative control siRNA), was used to demonstrate that transfection did not induce non-specific effects on gene expression. In miR-155 functional studies, cells were transfected with the hsa-miR-155 anti-miR oligo to inhibit the activity of endogenous miR-155. In parallel, cells were transfected with the FAM-labeled miR negative control oligo as a non-targeting negative control and to monitor transfection efficiency. All oligos were obtained from Ambion.
Statistical analysisDescriptive statistical analyses were conducted. For each set of experiments, values are reported as the mean±SD. Data were analyzed with Student's t test, and statistical significance was defined as p<0.05.
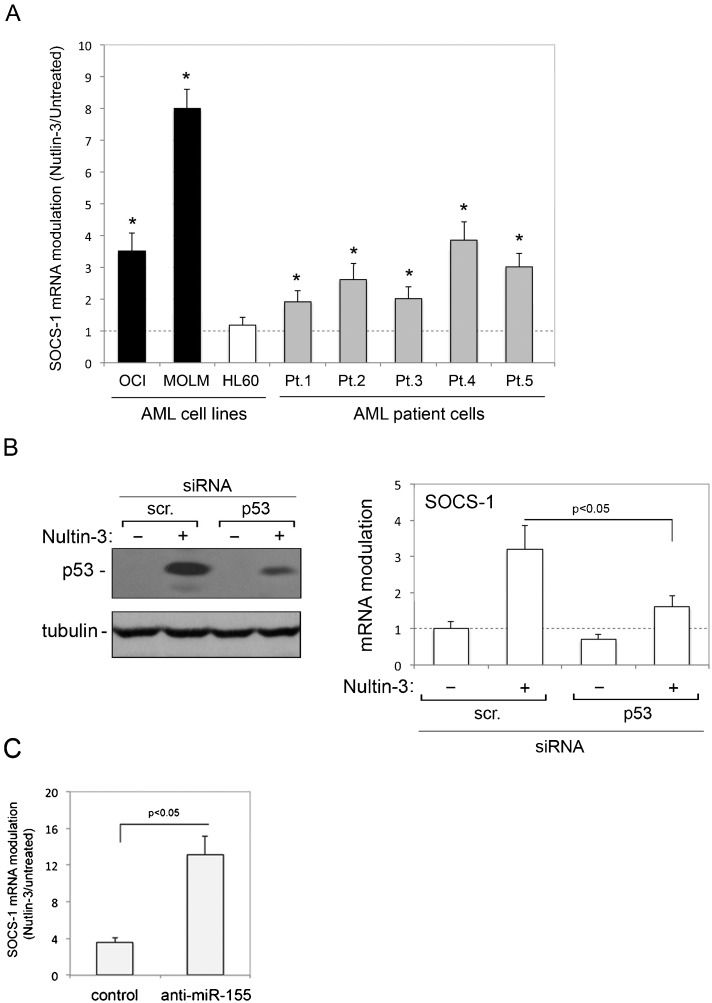
RESULTSUpregulation of SOCS-1 expression in p53wild-type AML cells by Nutlin-3In the first group of experiments, we investigated whether Nutlin-3 affected the expression of SOCS-1 mRNA in AML cells. For this purpose, we assessed both patient (n = 5) AML blasts and myeloid cell lines, characterized by either p53wild-type (OCI and MOLM) or p53null (HL-60) status. As reported in Table1, Nutlin-3 induced significant cytotoxicity in all patient AML blast cultures and p53wild-type myeloid cell lines but not in p53null HL-60 cells, which is consistent with the ability of Nutlin-3 to induce apoptosis and cell cycle arrest (2,4). Moreover, as shown in Figure1A), treatment with Nutlin-3 induced a significant (p<0.05) increase in SOCS-1 mRNA levels in all primary AML blasts and in p53wild-type OCI and MOLM cells but not in p53null HL-60 cells. In an attempt to also evaluate SOCS-1 protein, we analyzed cell lysates by the Western blot assay. However, under our experimental conditions, the commercially available antibodies did not allow for clear and reproducible protein detection (data not shown). The data shown in Figure1A) suggest but do not prove that the Nutlin-3-mediated upregulation of SOCS-1 in leukemic cells requires a functional p53 pathway. Therefore, to ascertain the role of p53 in Nutlin-3-induced upregulation of SOCS-1, we used predetermined optimal experimental conditions to transfect siRNA against p53 in OCI cells to specifically knock down p53 gene expression. As shown in Figure1B), p53 knock down counteracted the ability of Nutlin-3 to increase the level of p53 protein in OCI cells and significantly (p<0.05) counteracted the ability of Nutlin-3 to upregulate SOCS-1 expression. These data demonstrate that the ability of Nutlin-3 to transcriptionally upregulate SOCS-1 expression in myeloid leukemic cells is dependent on p53.
Effect of Nutlin-3 treatment on myeloid leukemic cell viability.
| Myeloidleukemic cells | Cell viability of Nutlin-3 treated cultures (% of untreated cultures)a) |
|---|---|
| OCI | 30±7 |
| MOLM | 6±3 |
| HL60 | 88±10 |
| Pt. 1 | 23±6 |
| Pt. 2 | 29±8 |
| Pt. 3 | 36±9 |
| Pt. 4 | 22±5 |
| Pt. 5 | 32±6 |
a Leukemic cells were exposed to Nutlin-3 (10 μM) and cell viability was analysed at 48 hours of treatment. Data are reported as means±SD of at least three independent experiments.
 cells were exposed to Nutlin-3 (10 μM). Levels of SOCS-1 mRNA were analyzed by quantitative RT-PCR. The results are expressed as the fold of increase in SOCS-1 modulation by Nutlin-3 after 24 hours of treatment with respect to the control untreated cultures (set to 1) (hatched line). Data are reported as the mean±SD of the results from at least three experiments, each performed in triplicate. In B, OCI cells were transfected with control scrambled (scr.) siRNA or p53 siRNA before treatment with Nutlin-3. p53 protein levels were analyzed by Western blot, and tubulin staining is shown as the loading control. Representative examples of Western blot results, from three independent experiments are shown. In parallel, levels of SOCS-1 mRNA were expressed as the fold increase with respect to the scrambled control transfected cultures. In C, OCI cells transfected with either hsa-miR-155 anti-miR oligo or miR negative control oligo were exposed to Nutlin-3 and SOCS-1 mRNA was expressed as the fold increase in modulation. Asterisks, p<0.05 with respect to the untreated cultures or to the scrambled control transfected cultures.")
Transcriptional upregulation of SOCS-1 by Nutlin-3 in AML cells. In A, AML cell lines and primary AML patient (Pt.) cells were exposed to Nutlin-3 (10 μM). Levels of SOCS-1 mRNA were analyzed by quantitative RT-PCR. The results are expressed as the fold of increase in SOCS-1 modulation by Nutlin-3 after 24 hours of treatment with respect to the control untreated cultures (set to 1) (hatched line). Data are reported as the mean±SD of the results from at least three experiments, each performed in triplicate. In B, OCI cells were transfected with control scrambled (scr.) siRNA or p53 siRNA before treatment with Nutlin-3. p53 protein levels were analyzed by Western blot, and tubulin staining is shown as the loading control. Representative examples of Western blot results, from three independent experiments are shown. In parallel, levels of SOCS-1 mRNA were expressed as the fold increase with respect to the scrambled control transfected cultures. In C, OCI cells transfected with either hsa-miR-155 anti-miR oligo or miR negative control oligo were exposed to Nutlin-3 and SOCS-1 mRNA was expressed as the fold increase in modulation. Asterisks, p<0.05 with respect to the untreated cultures or to the scrambled control transfected cultures.
The recent data demonstrating that Nutlin-3 downregulates miR-155 in primary B-CLL (8), along with a key role for miR-155 in regulating SOCS-1 expression (9,31,32), together supported the investigation of miR-155 knock down on SOCS-1 expression in response to Nutlin-3 in AML cell models. For this purpose, OCI cells were transfected either with the hsa-miR-155 anti-miR oligo to inhibit the activity of endogenous miR-155 or with the miR negative control oligo before exposure to Nutlin-3. Treatment with Nutlin-3 increased the levels of SOCS-1 mRNA in control (miR negative control oligo) transfected cells, and this effect was enhanced in cells transfected with anti-miR-155 (Figure1C). Although these experiments did not address a potential direct link between Nutlin-3 and miR-155, which also acts as oncomiR in AML (33,34), they confirm an important role for miR-155 in SOCS-1 transcriptional modulation and show that anti-miR-155 enhances the ability of Nutlin-3 to upregulate SOCS-1 mRNA expression in AML cells.
Because SOCS-1 is known to affect the intracellular signaling pathways downstream of several cytokines and one of its major targets is represented by the inhibition of the JAK/STAT-3 pathway (10,11), we analyzed the functionality of Nutlin-3-mediated induction of SOCS-1 by assessing the levels of phosphorylated STAT-3. For this purpose, leukemic cells were exposed to Nutlin-3 (10 μM), and at different time points, the phosphorylation pattern of the main intracellular pathways was evaluated by a multiplex assay. Among the activated pathways (Figure2), phosphorylation levels of both ERK1/2 and p38, although characterized by different baseline kinetics, were unaffected by Nutlin-3 exposure (Figure2A). Of interest, STAT-3 phosphorylation, characterized by a progressive increase over time in untreated cultures, was significantly inhibited after 24 hours of Nutlin-3 treatment, as further confirmed by Western blot analysis (Figure2B). Overall, these results are consistent with those of a previous study showing that ERK1/2 is not affected by SOCS-1 (35), and they validate the ability of SOCS-1 to downregulate the JAK/STAT-3 pathway upon Nutlin-3 treatment of leukemic cells.
, and phosphoprotein levels were analyzed in cell lysates by the Multiplex assay at the indicated time points. Phosphoprotein levels are expressed as the median fluorescence intensity (MFI). Data are reported as the mean±SD. In A, phosphorylation levels of ERK1/2 and p38 in untreated and Nutlin-3-treated cultures are shown. In B, phosphorylation levels of STAT-3 in untreated and Nutlin-3-treated cultures, analyzed with the Multiplex assay and validated by Western blot analysis, are shown. Asterisks, p<0.05 with respect to the untreated cultures.")
Phosphorylation patterns of Nutlin-3-treated AML cells. Leukemic cells were exposed to Nutlin-3 (10 μM), and phosphoprotein levels were analyzed in cell lysates by the Multiplex assay at the indicated time points. Phosphoprotein levels are expressed as the median fluorescence intensity (MFI). Data are reported as the mean±SD. In A, phosphorylation levels of ERK1/2 and p38 in untreated and Nutlin-3-treated cultures are shown. In B, phosphorylation levels of STAT-3 in untreated and Nutlin-3-treated cultures, analyzed with the Multiplex assay and validated by Western blot analysis, are shown. Asterisks, p<0.05 with respect to the untreated cultures.
Because SOCS-1 plays a role in different cell types (10), we analyzed whether Nutlin-3 was able to also modulate SOCS-1 in primary normal cells, such as normal macrophages, endothelial cells, and bone marrow MSCs, which represent the relevant cytotypes present in the bone marrow microenvironment. As shown in Figure3, Nutlin-3 did not significantly modulate SOCS-1 expression in any of the primary cell types investigated, suggesting that the ability of Nutlin-3 to upregulate SOCS-1 was confined to p53wild-type malignant AML cells (Figure3).
. Levels of SOCS-1 mRNA were analyzed by quantitative RT-PCR. The results are expressed as the fold increase in SOCS-1 modulation by Nutlin-3 after 24 hours of treatment with respect to the control untreated cultures (set to 1) (hatched line). Data are reported as the mean±SD of the results from at least three experiments, each performed in triplicate.")
Lack of transcriptional modulation of SOCS-1 by Nutlin-3 in normal cells. Normal adherent PBMC, endothelial cells, and MSC were exposed to Nutlin-3 (10 μM). Levels of SOCS-1 mRNA were analyzed by quantitative RT-PCR. The results are expressed as the fold increase in SOCS-1 modulation by Nutlin-3 after 24 hours of treatment with respect to the control untreated cultures (set to 1) (hatched line). Data are reported as the mean±SD of the results from at least three experiments, each performed in triplicate.
Previous findings indicated that SOCS-1 may function as a tumor suppressor and that it is frequently found to be silenced in hematopoietic malignancies. In this context, we have demonstrated that a novel non-genotoxic activator of the p53 pathway, Nutlin-3, is able to significantly upregulate SOCS-1 expression in primary AML blasts as well as in p53wild-type myeloid OCI and MOLM cell lines, but not in p53null HL-60 cells. In addition, we have demonstrated that the upregulation of SOCS-1 was functional because this upregulation resulted in a significant downregulation of STAT-3 phosphorylation levels. It is important to note that the activation of STAT-3 has been frequently reported in both primary human acute leukemia and leukemic cell lines (36), as well as that STAT-3 represents a major molecular target of SOCS-1 (10,11). However, no modulation of SOCS-1 was observed in primary normal macrophages, endothelial cells, or bone marrow MSC, suggesting that Nutlin-3 selectively upregulated SOCS-1 in malignant cells but not in normal cells of the bone marrow microenvironment. To date, we do not have an explanation for this differential activity of Nutlin-3 in myeloid leukemia cells and primary normal cells. We cannot exclude the possibility that Nutlin-3-mediated upregulation of SOCS-1 is also dependent on its ability to arrest cell cycle progression in highly cycling malignant cells, as some experimental observations (generated by blocking the cell cycle with different pharmacological agents) potentially support this hypothesis. However, these data are still too preliminary to draw any major conclusions. Additionally, a cycle-related effect of Nutlin-3 in upregulating SOCS-1 was also suggested by the more robust SOCS-1 induction in leukemic cell lines (3- to 8-fold) compared with primary AML (2- to 4-fold), a result also previously reported for primary B-CLL cells (2- to 4-fold; 9). Thus, further investigation is clearly needed to elucidate the molecular mechanisms underlining the differential behavior of Nutlin-3 with respect to SOCS-1 induction in normal cells compared with malignant cells.
A second interesting finding of our study was the identification that the Nutlin-3-mediated upregulation of SOCS-1 was potentiated by a concomitant downregulation of miR-155, which is known to act as an oncomiR in AML (33,34). Thus, SOCS-1 represents a potential common target between Nutlin-3 and antago-miR-155.
In conclusion, our data demonstrate that Nutlin-3, a small molecule with great therapeutic potential in hematological malignancies (37,38), upregulates the SOCS-1 pathway in leukemic cells but apparently not in normal quiescent cells. Due to the role of SOCS-1 as an oncosuppressor, therapeutic combinations based on Nutlin-3 warrant further investigations with regard to the treatment of AML.
AcknowledgmentsThis work was supported by the Regione Friuli Venezia Giulia - AITT Project.
AUTHOR CONTRIBUTIONSTisato V, Norcio A, Celeghini C, Gonelli A, and Milani D performed the experiments. Tisato V and Norcio A analyzed the data. Secchiero P designed the study and wrote the manuscript. All authors have read and approved the manuscript.
No potential conflict of interest was reported.