







Las pruebas genéticas (PG) proporcionan resultados que son para toda la vida y que tienen implicaciones no solo para el individuo sino para la familia, debiendo ir siempre acompañadas de consejo genético. Una de las herramientas que contribuye a que las PG se desarrollen en un entorno apropiado es el documento de consentimiento informado (DCI). El consentimiento informado (CI) tomado por el facultativo que prescribe la prueba es un medio para garantizar el acceso libre e informado y para que la persona entienda el propósito de la misma y las implicaciones de los resultados, así como para garantizar su derecho a recibir el consejo genético, ya que le da la oportunidad de hacer preguntas y también de manifestar su derecho a decidir qué información quiere conocer y cuál no quiere conocer en cualquier momento del proceso.
En el presente trabajo se hace una reflexión sobre los principios éticos que fundamentan la toma del CI para la realización de PG y cómo nuestra sociedad ha plasmado en forma de regulaciones legales la protección de los valores éticos y de la dignidad y derechos fundamentales de los individuos con relación a las PG y a la información genética: Ley 14/2007 de 3 de julio de Investigación Biomédica. Como conclusiones se proponen 3 modelos de DCI: para la realización de PG en el contexto asistencial, con un consentimiento adicional en el caso de que el laboratorio clínico desee guardar las muestras excedentes para futuros usos en investigación y para un proyecto de investigación que incluya PG.
The results obtained in genetic tests are valid for the whole life. They are important, not only for the individual, but also for the family. Genetic counselling must be an integral part of the genetic testing process. An important tool that must be used in order that genetic testing is performed properly is the informed consent document. This informed consent, obtained by the physician who requests the genetic tests, is a resource to ensure that the individual voluntarily agrees and understands their purposes, as well as the consequences of the results. It emphasises the rights to receive genetic counselling, and gives the opportunity to ask questions. It also mentions the right to choose what information the subject wishes or does not wish to know.
This article considers the ethical principles that justify the informed consent and its inclusion in legal regulations in order to protect fundamental human rights concerning genetic testing and genetic information, such as Spanish law 14/2007 on Biomedical Research. As conclusions, 3 models of informed consent are proposed: one for genetic testing for health reasons; an additional consent in cases where the clinical laboratory wants to store the remaining samples for future uses in biomedical research, and for clinical trials that including genetic tests.
La toma del consentimiento informado (CI) voluntario y competente en el ámbito asistencial tiene lugar antes de llevar a cabo una prueba invasiva y de emprender determinadas intervenciones sanitarias que, aunque comporten beneficios, impliquen también riesgos potenciales o resultados inciertos o cuando se rehúsan cuidados médicos habituales. En este ámbito, el CI a veces contiene elementos para proteger a los profesionales frente a reclamaciones legales.
El CI es asimismo un paso indispensable para la participación en un proyecto de investigación, después de recibir información sobre los procedimientos y sus beneficios y riesgos. Su principal objetivo ético es proteger a la persona participante frente a daños físicos, psicológicos y sociales (estigmatización, exclusión social). Vale la pena recordar en este sentido que la declaración de Helsinki (1964/1975/2000) remarca que las consideraciones sobre el bienestar de los individuos participantes deben prevalecer sobre cualquier interés o beneficio colectivo de la sociedad o de la ciencia. El consentimiento informado es además una garantía para promover la confianza del público en la investigación y en los investigadores.
Son elementos fundamentales de la toma del CI:
- a)
El documento de consentimiento informado (DCI).
- b)
Quién toma el consentimiento y cómo lo obtiene, incluyendo el proceso de información; la forma de comunicación, cuidando el lenguaje médico científico y siendo culturalmente sensible; evitando la coerción; tomándose el tiempo necesario para la verificación de que se ha comprendido la información y el alcance del consentimiento.
- c)
Cómo y quién asume la responsabilidad en el caso de personas que no tienen capacidad para consentir adecuadamente.
- d)
Cómo se registra y archiva el DCI.
Naturalmente el DCI y el proceso de toma de consentimiento pueden ser muy distintos cuando se realizan para someterse a una prueba para un diagnóstico diferencial en el ámbito de la asistencia sanitaria habitual o cuando se toman para participar en un proyecto de investigación biomédica.
Principios éticos que fundamentan el documento del consentimiento informado para la realización de pruebas genéticasLas pruebas genéticas (PG) proporcionan resultados que son para toda la vida, tienen implicaciones familiares y generacionales y pueden ser determinantes para la toma de decisiones en el diagnóstico prenatal. La información genética que de ellas se obtiene a menudo es predictiva y el énfasis no suele estar en el tratamiento, es privada y compleja, suele ser grave y comporta dilemas éticos. Puede ser identificativa y dar lugar a hallazgos inesperados. Aun reconociendo que la información genética no es intrínsecamente excepcional según la concepción actual de salud y enfermedad1–4, los aspectos éticos relativos a las pruebas genéticas y la información genética que con ellas se obtiene son muy relevantes.
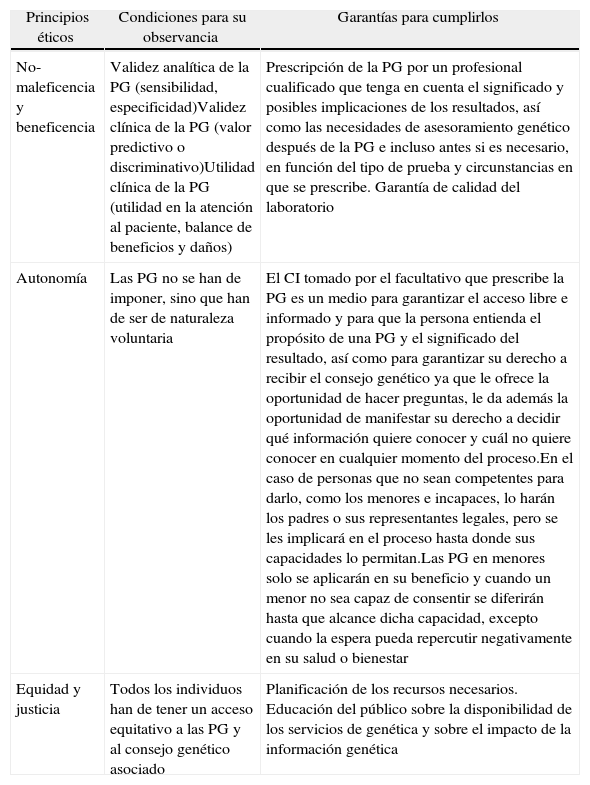
Haciendo una aproximación desde la perspectiva de la ética normativa y la fuente tradicional principialista de guía ética en medicina, se han resumido esquemáticamente en la tabla 1 las condiciones mínimas que deben satisfacerse, con relación a las PG, para la observancia de los 4 principios éticos básicos en biomedicina5 así como los medios para garantizar su cumplimiento.
Condiciones mínimas que deben satisfacerse, con relación a las pruebas genéticas para la observancia de los principio éticos básicos en biomedicina
| Principios éticos | Condiciones para su observancia | Garantías para cumplirlos |
| No-maleficencia y beneficencia | Validez analítica de la PG (sensibilidad, especificidad)Validez clínica de la PG (valor predictivo o discriminativo)Utilidad clínica de la PG (utilidad en la atención al paciente, balance de beneficios y daños) | Prescripción de la PG por un profesional cualificado que tenga en cuenta el significado y posibles implicaciones de los resultados, así como las necesidades de asesoramiento genético después de la PG e incluso antes si es necesario, en función del tipo de prueba y circunstancias en que se prescribe. Garantía de calidad del laboratorio |
| Autonomía | Las PG no se han de imponer, sino que han de ser de naturaleza voluntaria | El CI tomado por el facultativo que prescribe la PG es un medio para garantizar el acceso libre e informado y para que la persona entienda el propósito de una PG y el significado del resultado, así como para garantizar su derecho a recibir el consejo genético ya que le ofrece la oportunidad de hacer preguntas, le da además la oportunidad de manifestar su derecho a decidir qué información quiere conocer y cuál no quiere conocer en cualquier momento del proceso.En el caso de personas que no sean competentes para darlo, como los menores e incapaces, lo harán los padres o sus representantes legales, pero se les implicará en el proceso hasta donde sus capacidades lo permitan.Las PG en menores solo se aplicarán en su beneficio y cuando un menor no sea capaz de consentir se diferirán hasta que alcance dicha capacidad, excepto cuando la espera pueda repercutir negativamente en su salud o bienestar |
| Equidad y justicia | Todos los individuos han de tener un acceso equitativo a las PG y al consejo genético asociado | Planificación de los recursos necesarios. Educación del público sobre la disponibilidad de los servicios de genética y sobre el impacto de la información genética |
Otros derechos a proteger son el derecho a no saber, el derecho a la confidencialidad de los datos personales y especialmente de los genéticos, el derecho a la no discriminación y el de decidir sobre la custodia de las muestras y el posible uso futuro tanto de los datos como de las muestras en investigación biomédica.
El establecimiento de líneas guía, recomendaciones y declaraciones referentes a las PG y el consejo genético asociado a las mismas, así como las regulaciones legales son una realidad en el ámbito internacional6. España es uno de los países firmantes del convenio impulsado por el Consejo de Europa para la protección de los derechos humanos y la dignidad del ser humano respecto a las aplicaciones de la biología y de la medicina, conocido también como Convenio de Oviedo (1997). Más recientemente el Consejo de Europa aprobó el protocolo adicional a dicho convenio que hace referencia específica a las PG con propósitos sanitarios7.
Por coherencia con la protección de estos derechos fundamentales, España promulgó la Ley 14/2007, de 3 de Julio de Investigación Biomédica (LIB) que, entre otras cuestiones importantes, establece el marco jurídico en el que se ha de situar la realización de análisis genéticos con cualquier finalidad, incluida la asistencial. Recomendamos su lectura completa, y de manera específica para el presente tema la del Título V: Análisis genéticos, muestras biológicas y biobancos.
La LIB contempla la voluntariedad de acceso a las PG y su realización bajo consentimiento informado (expreso y específico, es decir, por escrito), la realización bajo criterios de pertinencia, el compromiso del consejo genético, el derecho a la información y el derecho a no ser informado, la advertencia de las implicaciones que puede tener para sus familiares y la conveniencia de que se les transmita la información, la preservación de la confidencialidad y la protección de los datos.
Cuando una PG se lleva a cabo como intervención sanitaria dentro de un proceso asistencial, su validez analítica y clínica y su utilidad clínica habrán sido previamente demostradas. Cuando se realiza en el contexto de un proyecto de investigación, estas características pueden formar parte de la hipótesis de trabajo y las incógnitas no se despejarán hasta que finalice el proyecto, por lo tanto el CI tendrá elementos distintivos en ambos casos. En las 2 situaciones, la muestra biológica remanente puede ser destruida al final del proceso o ser conservada con propósito de investigaciones ulteriores y este hecho requiere un tratamiento específico en ambos tipos de consentimiento.
A continuación se expone brevemente: 1) qué se considera PG y por lo tanto requiere consentimiento informado, 2) elementos que debe contener el CI para la realización de PG en el ámbito asistencial, 3) elementos del consentimiento para guardar y disponer del material biológico sobrante con propósitos de investigaciones futuras, y 4) elementos que debe contener el CI cuando las PG forman parte de un proyecto de investigación.
Qué se considera prueba genética y por lo tanto requiere consentimiento informadoPG o análisis genético son los términos empleados principalmente para pruebas analíticas realizadas en laboratorios de genética: citogenética, genética molecular y genética bioquímica, como parte de los servicios de genética.
El término PG es complejo y se usa a veces con significados distintos, pero su definición es imprescindible cuando va a ser objeto de legislación, recomendaciones de política sanitaria o establecimiento de líneas guía profesionales. Debido a ello, definir PG era uno de los objetivos del proyecto EUROGENTEST. En el trabajo de Pinto-Basto et al.8 se describe cómo se abordó el tema; el resultado fue que a pesar de que se reconoció por amplia mayoría la necesidad de una definición de consenso, se estimó remota la posibilidad de alcanzarla, sin embargo dos tercios de los participantes estuvieron de acuerdo en que la información que debía quedar cubierta por el término «prueba genética» sería la concerniente a ADN, cromosomas y genética bioquímica; un 38% añadió además cualquier prueba que proporcionase información genética inequívoca independientemente de su naturaleza.
La Declaración Internacional sobre Datos Genéticos Humanos de la UNESCO9 define como PG «un procedimiento para detectar la presencia o ausencia, o cambio en un gen particular o cromosoma, incluyendo una prueba indirecta para un producto génico u otros metabolitos específicos que sean primariamente indicativos de un cambio genético específico» y como dato genético humano la «información acerca de características hereditarias de los individuos obtenidas mediante análisis de ácidos nucleicos o por otros análisis científicos». El Protocolo Adicional a la Convención de Derechos Humanos y Biomedicina del Consejo de Europa7, concerniente a las pruebas genéticas con propósitos sanitarios las define como «análisis cromosómicos, de DNA o RNA y análisis de otros elementos que faciliten la obtención de información equivalente a la obtenida con los métodos anteriores».
La definición de análisis genético que da la LIB en su Título I, artículo 3 se inspira en las anteriores: «Procedimiento destinado a detectar la presencia, ausencia o variantes de uno o varios segmentos de material genético, lo cual incluye las pruebas indirectas para detectar un producto génico o un metabolito específico que sea indicativo ante todo de un cambio genético determinado». Para una revisión de las definiciones de PG en documentos europeos y otros documentos legales ver Varga et al.10 y Sequeiros et al.11.
La inclusión en la definición de PG del producto del gen y de metabolitos patognomónicos, es decir de lo que constituye el ámbito de la genética bioquímica, es crucial para las enfermedades metabólicas hereditarias porque garantiza el asesoramiento genético en el momento adecuado y la protección de los resultados como información genética.
Las técnicas analíticas de las PG pueden emplearse para diagnosticar o seguir alteraciones genéticas no constitucionales o familiarmente heredadas, sino sobrevenidas o circunstanciales a nivel de líneas celulares, tejidos, fluidos biológicos, por ejemplo enfermedades metastáticas no familiares (somáticas), o estudio de material genético de agentes infecciosos. Por ello vale la pena remarcar que el anteriormente citado Protocolo Adicional a la Convención de Derechos Humanos y Biomedicina del Consejo de Europa concerniente a las Pruebas Genéticas con Propósitos Sanitarios7 establece al inicio del documento que «el protocolo aplica a pruebas que se llevan a cabo con propósitos sanitarios, implicando muestras biológicas de origen humano y dirigidas específicamente a identificar las características genéticas de una persona que son heredadas o adquiridas precozmente durante el desarrollo prenatal (y a las cuales se referirá mas adelante como «PG»), es decir se aplica a características constitucionales. En la LIB no se especifica, y aunque el conjunto del redactado tiene el mismo sentido que el Protocolo del Consejo de Europa, puede haber suscitado alguna duda.
El documento de consentimiento informado para la realización de pruebas genéticas en el ámbito asistencial. Elementos que debe contenerLa LIB especifica que el sujeto debe recibir información por escrito sobre varios puntos: finalidad del análisis genético; lugar de realización de la PG y destino de la muestra biológica al final del mismo; personas que tendrán acceso a los resultados de los análisis; advertencia sobre la posibilidad de descubrimientos inesperados y su posible trascendencia, así como su facultad de tomar una posición en relación con recibir su comunicación; advertencia de la implicación que puede tener la información obtenida para sus familiares y la conveniencia de que se le transmita; el compromiso de suministrar consejo genético.
Cuando la PG se practica en personas que no sean capaces de dar el consentimiento (menores o personas con sus capacidades disminuidas), lo harán los padres o representantes legales, pero debe implicárseles en el proceso de decisión hasta donde sus capacidades lo permitan. Debe tenerse en cuenta además que solo se llevarán a cabo si es en su propio beneficio. Cuando, de acuerdo con la ley, un menor no tenga capacidad de consentir, se diferirá la PG hasta que alcance dicha capacidad, a menos que ello fuese en detrimento de su salud o bienestar12–15. A partir de los 12 años, es conveniente que en el consentimiento se recoja la firma del menor, junto a la de sus padres o tutores.
En el caso de PG en muestras de personas fallecidas, para esclarecer o complementar información relevante para la salud de un familiar vivo, el consentimiento lo otorgarán los padres, o los hijos o representantes legales en función de la situación. La necesidad del estudio de familiares de grado de parentesco lejano suele ser excepcional y además es difícil que se hayan conservado las muestras, pero si así fuese, el consentimiento debería solicitarse al familiar más próximo en el árbol genealógico.
Cuando el paciente es un feto la situación es la misma, si bien en este caso habrá un consentimiento adicional para la prueba invasiva que pueda comportar riesgos físicos específicos.
Cuando la PG forme parte de un programa de salud pública de cribado de población, los documentos de consentimiento formarán parte del protocolo del programa. Los cribados de población tienen connotaciones éticas específicas16 y si bien no constituyen el objeto del presente artículo, vale la pena remarcar que el programa debe ser revisado y aprobado por un Comité de Ética, hecho que se recoge también en nuestra LIB.
En el caso de que el laboratorio desee guardar las muestras remanentes para futuros usos en investigación, se firmará un consentimiento adicional que contendrá elementos específicos. Ver el siguiente apartado.
El documento de consentimiento informado para guardar y disponer del material biológico excedente de procesos asistenciales con propósitos de investigaciones futuras. Elementos que debe contenerDeberá contener como mínimo los elementos siguientes: las líneas generales de investigación para las que van a ser usadas las muestras; que el material biológico pasará a formar parte de las colecciones del centro de acuerdo con las condiciones establecidas por la LIB; que las investigaciones se realizarán en las instalaciones del centro y en aquellas otras instituciones de investigación colaboradoras; que todo investigador que solicite la utilización de las muestras deberá disponer de la aprobación previa del Comité de Ética de Investigación (CEI); que la identificación de las muestras será codificada; que los datos personales asociados serán siempre confidenciales y procesados de acuerdo con la Ley Orgánica de Protección de Datos (LOPD); que podrá ejercer sus derechos de acceso, rectificación y cancelación del fichero, obtener información del uso de las muestras y revocar el consentimiento que presta en cualquier momento y sin necesidad de dar explicaciones; que la cesión de muestras que realiza es gratuita y altruista, por ello no obtendrá retribución económica alguna ni tendrá derecho sobre posibles beneficios comerciales como resultado de las investigaciones realizadas; que si de la investigación con sus muestras se obtuviesen beneficios relevantes para su salud o la de sus familiares el CEI habilitará los medios oportunos para contactar con él y ofrecerle la posibilidad de conocer dicha información (los datos que figuren en su historial clínico) pero se respetará su derecho a decidir que no se le comuniquen los resultados de la investigación en que hayan sido utilizadas sus muestras; que si no desea que sus muestras sobrantes sean utilizadas para la investigación biomédica, en ningún caso repercutirá negativamente en el cuidado asistencial que recibirá; que tiene derecho a saber en qué investigaciones se han utilizado sus muestras17. Ver también la LIB y el Real Decreto 17/16/2011 de 18 de noviembre.
El consentimiento informado cuando las pruebas genéticas forman parte de un proyecto de investigación. Elementos que debe contenerEl CI en investigación se apoya en 3 principios éticos fundamentales: respeto por las personas (autonomía), beneficencia y justicia distributiva. Se estableció como resultado de los principios básicos formulados para la investigación en seres humanos en el Código de Nuremberg, estando también presente en la Declaración de Helsinki y en el Informe Belmont. Ver también las líneas guía de la CIOMS18.
Cuando el CI se toma para un proyecto de investigación tanto si incluye PG como si no, debe informar de: la naturaleza y objetivos experimentales del estudio; justificación de la invitación a participar y su naturaleza voluntaria; molestias y riesgos si los hubiera; incertidumbre de los resultados; posibles beneficios para otros y para la ciencia; confidencialidad de los registros; cómo contactar para preguntar sobre la investigación y también en caso de daños; derecho a revocar el consentimiento y retirarse; derecho a recibir cuidados sanitarios sin restricciones, incluso si se retira; derecho a decidir si desea o no recibir información de los resultados que le conciernan y quién, cuándo y cómo le informará; posibilidad del uso comercial de los resultados de la investigación y que el sujeto fuente no recibirá beneficio económico si lo hubiera; publicación de los datos agregados y en caso de que se identifique a la persona, solo con su consentimiento; lugar de realización y destino de la muestra biológica al final del mismo; personas que tendrán acceso a los resultados; garantía de que el proyecto ha sido aprobado por un Comité de Ética. Cabría también preguntar si acepta que en caso de ser necesaria alguna muestra adicional, el centro podría ponerse en contacto de nuevo con él para solicitarle nuevamente su colaboración.
Si el proyecto conlleva la realización de PG debe incluir además una advertencia sobre el derecho a recibir asesoramiento genético, la posibilidad de descubrimientos inesperados y su posible trascendencia, así como su facultad de tomar una posición en relación con recibir su comunicación. Ver LIB y Wertz et al.19.
La investigación en genética y sus aspectos éticos requeriría un artículo aparte, incluyendo además aspectos específicos para la aplicación de las nuevas técnicas de secuenciación masiva y la obtención del genoma personal20–23, el presente artículo es una llamada de atención únicamente a la inclusión de PG clásicas dentro de un proyecto de investigación genérico.
ConclusionesEn función de las consideraciones de los apartados anteriores y el debate generado en torno al seminario «El documento de consentimiento informado: para pruebas genéticas con finalidades asistenciales. Para pruebas genéticas en proyectos de investigación» del XXVI Congreso de la Asociación Española de Genética Humana (AEGH) celebrado en Murcia en marzo de 2011, la Comisión de Ética de la AEGH ha elaborado los siguientes modelos de CI, para la realización de PG con fines asistenciales (anexo 1), para la retención y uso en investigación de las muestras excedentes de los procesos asistenciales (anexo 2) y para un proyecto de investigación que contenga PG (anexo 3). La Junta directiva de la AEGH ha revisado y aprobado asimismo dichos documentos.
Estos modelos se proponen como una herramienta que puede ser útil para que las personas que prescriben y realizan PG preparen su modelo de CI para presentarlo a la revisión y aprobación por el Comité de Ética de su institución, bien entendido que deberán tener en cuenta las observaciones/modificaciones/cambios que dicho comité considere oportunos. Los modelos sugeridos no tienen ninguna validez, ni deben ser usados, sin su revisión y aprobación por un Comité de Ética competente.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
Agradecemos profundamente a la Dra. Pilar Nicolás, de la Cátedra Interuniversitaria de Derecho y Genoma Humano (Interuniversity Chair in Law and the Human Genome) Universidad del País Vasco (Euskal Herriko Uniberstitatea), Universidad de Deusto, la discusión y observaciones acerca de los modelos de DCI elaborados por la Comisión de Ética de la AEGH ya que nos han sido muy útiles y los han mejorado notablemente. Agradecemos a la Junta directiva de la AEGH la revisión y aprobación de los modelos de DCI y las enriquecedoras deliberaciones que nos han suscitado. También agradecemos a los Dres. Antonio Matilla, del Hospital Universitari Germans Trías i Pujol y José Antonio López-Guerrero de la Fundación Valenciana de Oncología, el envío de sus modelos de CI para compartir experiencias. Se ha tenido en cuenta, asimismo, el modelo de CI del Servicio de Bioquímica y Genética Molecular del Hospital Clínico de Barcelona.




