


la incidencia de las alteraciones del desarrollo de las estructuras osteocartilaginosas fetales es baja. Aunque su diagnóstico ecográfico es sencillo, especialmente en las osteocondrodisplasias letales, llegar a un diagnóstico etiológico es complicado. El objetivo del presente trabajo es presentar los hallazgos ecográficos y resultados perinatales de las malformaciones esqueléticas diagnosticadas en nuestro centro.
Material y métodosestudio descriptivo retrospectivo de serie de casos. Se incluyeron las malformaciones esqueléticas diagnosticadas prenatalmente en la Sección de Medicina Fetal del Hospital General Universitario Gregorio Marañón (noviembre de 2004-febrero de 2010). Los casos se clasificaron en defectos del esqueleto axial o radial, tanto focales aislados como cuadros generalizados de displasias óseas. Se analizaron la edad gestacional al diagnóstico, los defectos congénitos asociados y los resultados perinatales.
Resultadosdurante el período de estudio se registraron 56 casos. La edad gestacional media al diagnóstico fue de 23,7 semanas (rango 12-37; desviación estándar 7,25). Veintiséis (46,4%) se presentaron como malformación aislada; 14 (25%) asociadas a otras malformaciones, y 16 (28,6%) en el contexto de un síndrome polimalformativo. De los 29 (51,8%) casos en que se realizó estudio citogenético, en 10 (34,5%) se diagnóstico una cromosomopatía. En 40 casos (71,4%) el diagnóstico fue de anomalía focal del esqueleto radial; en 5 (8,9%), de anomalía del esqueleto axial, y en 14 (25%), de osteocondrodisplasia. Sólo en 11 casos se estableció un diagnóstico nosológico. La supervivencia en el período neonatal fue del 61,1% (33/54).
Conclusioneslas malformaciones esqueléticas se asocian frecuentemente a otros defectos congénitos, de difícil diagnóstico etiológico y con una alta tasa de resultado perinatal adverso.
The incidence of disorders in the development of foetal bone and cartilage structures is low. Although ultrasound diagnosis is easy, especially in lethal skeletal dysplasias, to reach an aetiological diagnosis can be difficult. The aim of this study was to present the ultrasound findings and perinatal outcomes of skeletal malformations diagnosed in our centre.
Material and methodsA retrospective descriptive study of case series. Skeletal malformations prenatally diagnosed in the Foetal Medicine Unit at the Gregorio Marañón General University Hospital (HGUGM) Madrid between November 2004 and February 2010 were included. Cases were classified as axial or radial skeletal defects, including isolated defects, as well as generalised skeletal dysplasias. Gestational age at diagnosis, presence of associated congenital defects and perinatal outcome were recorded.
ResultsA total of 56 cases were included during the study period. The mean gestational age at diagnosis was 23.7 weeks (range 11-36, standard deviation 7.25). Twenty-six (46.4%) occurred as an isolated malformation, 14 (25%) associated to other malformations, and 16 (28.6%) as a multiple malformation syndrome. Among the 29 (51.8%) cases in which a cytogenetic study was performed, 10 (34.5%) had a chromosomal abnormality. The diagnosis was a radial skeletal focal anomaly in 40 cases (71.4%); anomalies of the axial skeleton in 5 (8.9%); and osteochondrodysplasia in 14 (25%). A nosological diagnosis was established in only 11 cases. Neonatal survival was 61.1% (33/54).
ConclusionsSkeletal malformations are often associated with other congenital defects. Aetiological diagnosis may be extremely difficult and the rate of adverse perinatal outcomes is high.
Las anomalías esqueléticas son alteraciones de la formación o el desarrollo de las estructuras osteocartilaginosas, y comprenden un grupo de malformaciones muy heterogéneo desde un punto de vista fenotípico y genético. Su incidencia es baja, aproximadamente de 1/2.000-4.000 recién nacidos vivos. Pueden presentarse de forma aislada o bien asociadas a otro defecto congénito, ya sea una malformación estructural, cromosomopatía o síndrome polimalformativo.
El diagnóstico ecográfico de estas anomalías, sobre todo de las displasias esqueléticas letales, es sencillo pero no supone más que el punto de partida del diagnóstico prenatal1. Además, se deben realizar las pruebas complementarias necesarias, principalmente genéticas, para intentar establecer un diagnóstico etiológico que permita un tratamiento individualizado en función del pronóstico del caso. El pronóstico, principalmente en las osteocondrodisplasias y en el contexto de síndromes polimalformativos, es malo. En caso de interrupción legal del embarazo (ILE), fallecimiento intrauterino o muerte perinatal, debe completarse el estudio, en el que se incluyan una serie ósea y necropsia.
En el presente artículo se presenta un análisis descriptivo de las malformaciones esqueléticas diagnosticadas por ecografía, atendidas en la Sección de Medicina Fetal de nuestro centro.
Material y métodosSe ha realizado un estudio descriptivo retrospectivo de evaluación de una serie de casos.
Criterios de inclusiónLa muestra de pacientes seleccionada comprende los casos diagnosticados de anomalía esquelética mediante ecografía obstétrica, en el período comprendido entre noviembre de 2004 y febrero de 2010, en la Sección de Medicina Fetal de nuestro centro. Se incluyen las anomalías esqueléticas que afectan al esqueleto axial o radial diagnosticadas de forma aislada, así como los cuadros generalizados de displasias óseas.
Los datos prenatales se obtuvieron de la base de datos de defectos congénitos de la Sección de Medicina Fetal durante el período de estudio. Se recogió la edad materna, la edad gestacional (EG) en el momento del diagnóstico ecográfico, el tipo de gestación (única o gemelar con diagnóstico de corionicidad), el diagnóstico ecográfico principal, las anomalías asociadas (otras malformaciones estructurales, cromosomopatías o síndromes polimalformativos). Los resultados perinatales se obtuvieron de la historia clínica o, en caso de que el parto se atendiera en otro centro, por contacto telefónico. Se recogió la incidencia de ILE, fallecimiento intraútero y mortalidad perinatal; en los casos de gestaciones que siguieron su curso, se registró la EG al parto, vía del parto, peso del recién nacido y Apgar a los 5 minutos.
Se utilizó el paquete estadístico SPSS versión 16.0 para el análisis de los datos.
Clasificación de los casosLos casos se clasificaron en defectos del esqueleto axial (cráneo, tórax y columna vertebral) o radial (cintura escapular, pelviana y extremidades), tanto malformaciones focales aisladas, como cuadros generalizados de displasias óseas. Se elaboró una tabla con los diagnósticos realizados, tomada parcialmente de la clasificación de Swanson de 1983, para las malformaciones congénitas de las extremidades2. La clasificación de las osteocondrodisplasias se estableció de acuerdo con la letalidad del defecto y su localización. Esta clasificación se basa en la publicada por el International Working Group on Constitutional Diseases of Bone en 19973,4. Complementariamente, en los casos en los que fue posible, se utilizó la clasificación del Nosology Group of the International Skeletal Dysplasia Society (ISDS)5 que hace referencia a la etiología genética de estas alteraciones.
En función de la asociación a otros defectos congénitos, la anomalía esquelética se clasificó como aislada, asociada a otra malformación estructural o a cromosomopatía, o bien en el contexto de un cuadro polimalformativo.
ResultadosSe reunieron un total de 56 casos que satisfacían los criterios de inclusión del presente estudio.
La edad media materna fue de 31 años (rango, 15-47). Un 96,4% (54/56) de los casos se presentaron en gestaciones únicas y un 3,6% (2/56), en gestaciones gemelares bicoriales con afectación de un único gemelo. No hubo ningún caso de gestación gemelar monocorial. La EG media al diagnóstico fue de 23,7 semanas (rango 12-37; desviación estándar [DE] 7,25) y presentó una distribución bimodal, con dos picos en torno a las semanas 20 y 35.
En un 51,8% (29/56) de los casos se realizó estudio citogenético mediante biopsia corial, amniocentesis o funiculocentesis con resultado de 10 cariotipos patológicos (10/29; 34,5%) y un caso (1/29; 3,5%) de defecto monogénico (mutación homocigota Arg279Trp del gen SLC26A2, del transportador de sulfatos). Entre las cromosomopatías se diagnosticaron 5 trisomías 21 (5/10; 50%), 2 trisomías 18 (2/10; 20%), una trisomía 13 (1/10, 10%), una delección parcial en el brazo corto del cromosoma 5 (5p) (1/10; 10%) y una triploidía (1/10; 10%).
Respecto a las malformaciones estructurales asociadas, un 46,4% (26/56) de las alteraciones esqueléticas se presentó de forma aislada; un 25% (14/56), asociada a otro tipo de malformación, y un 28,6% (16/56), en el contexto de un síndrome polimalformativo.
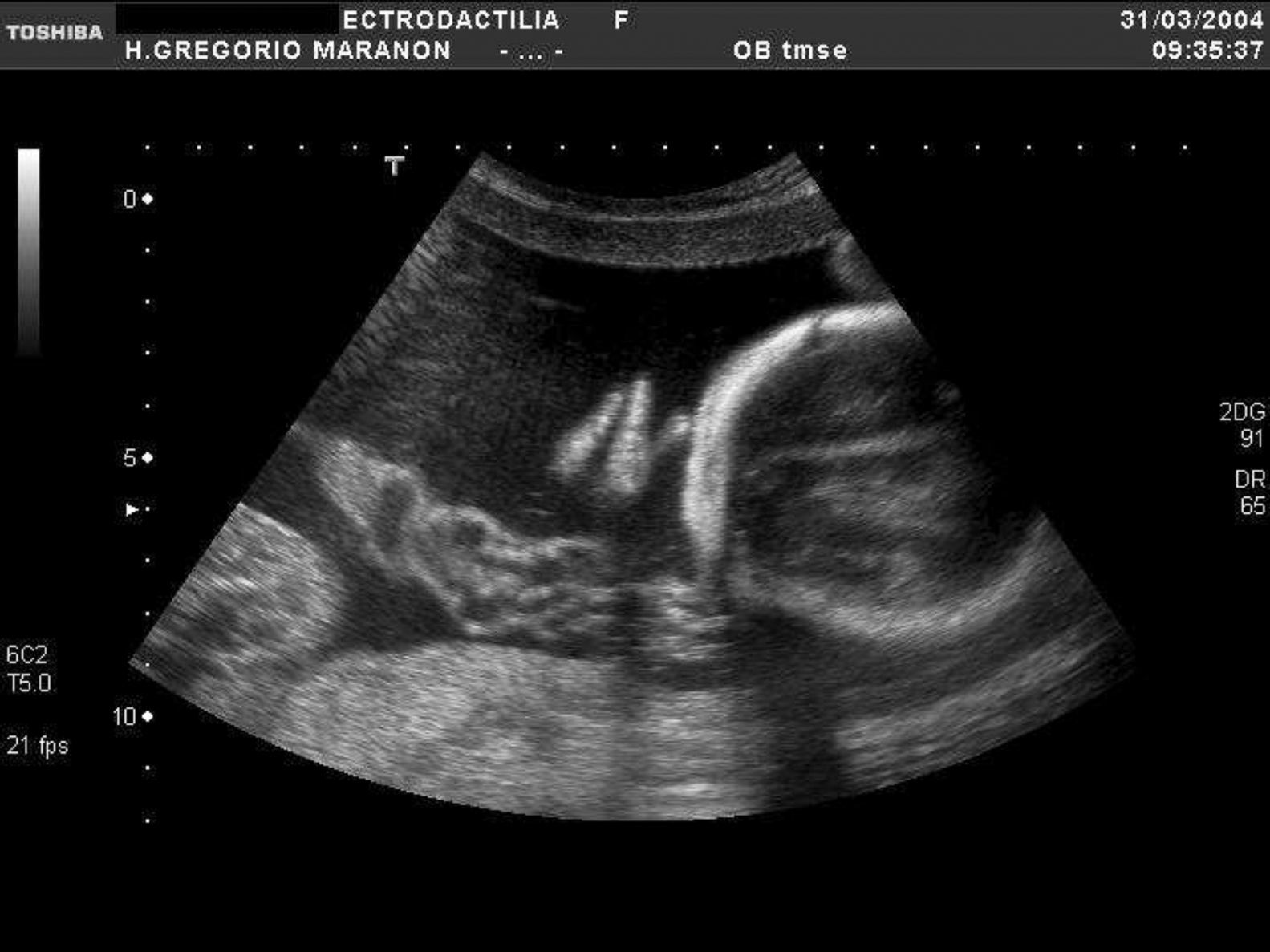
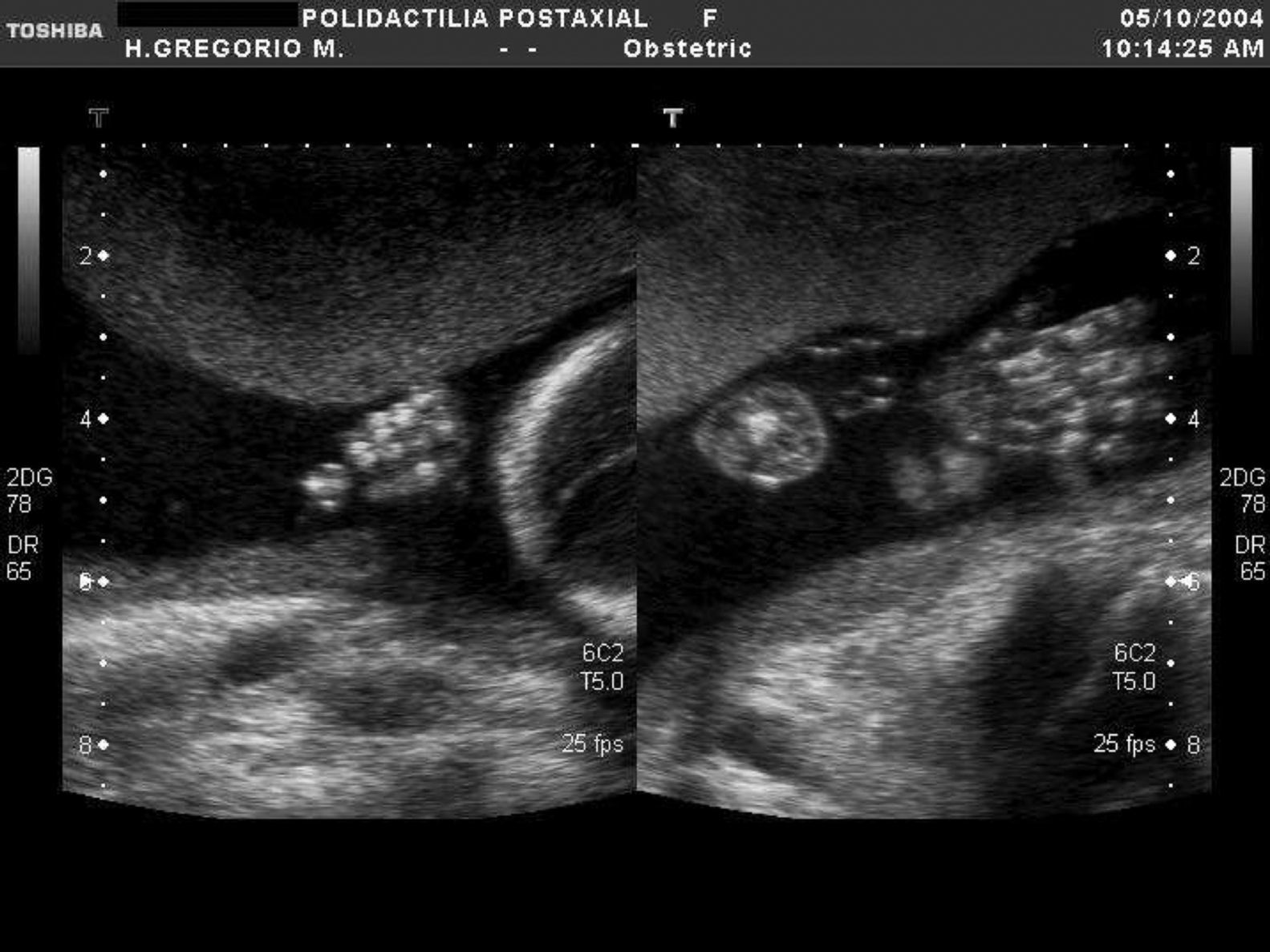
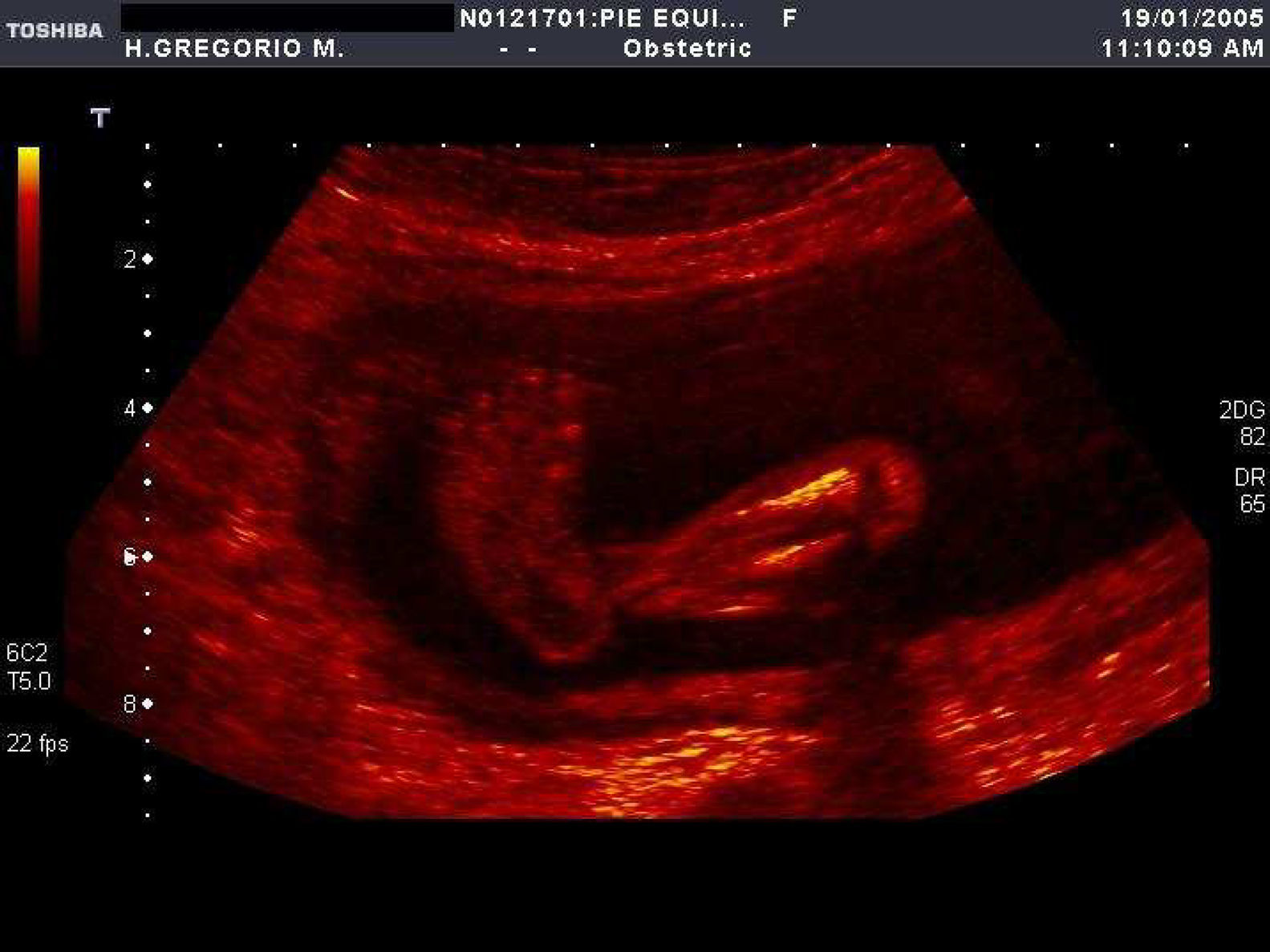
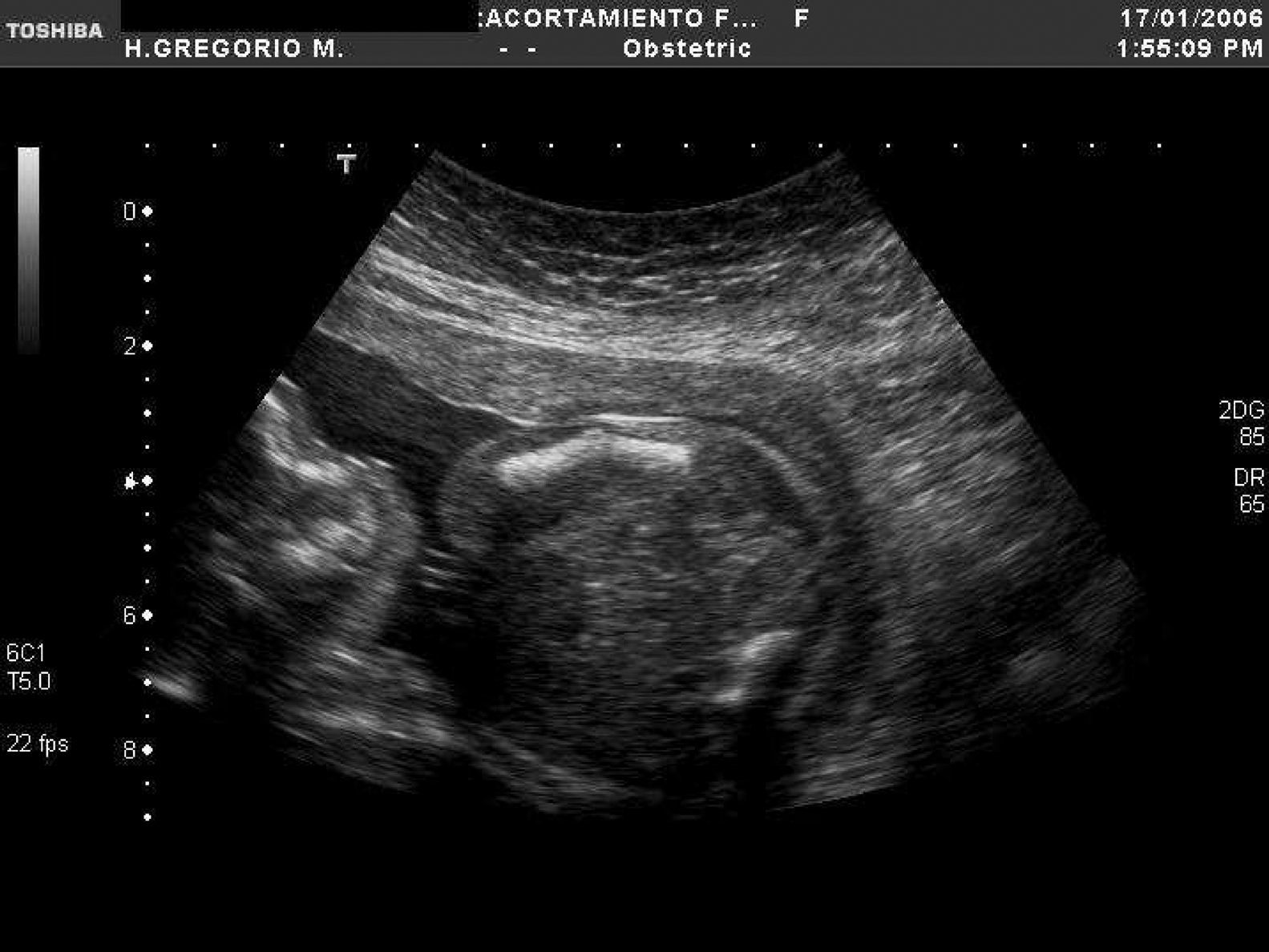
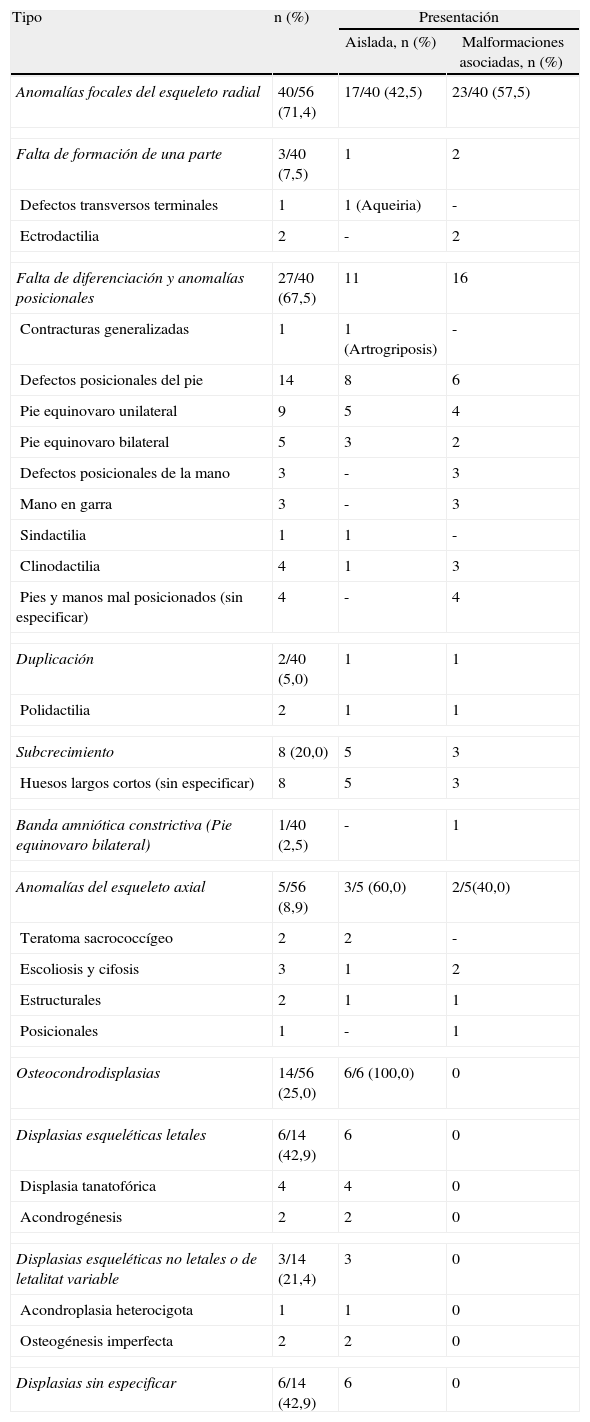
En la tabla 1 se expone el tipo de malformación esquelética detectada según la clasificación previamente descrita2. Se registraron 45 casos de anomalías esqueléticas focales, distribuidas de forma muy heterogénea. Veinticinco de ellas se presentaron en el contexto de síndromes polimalformativos. Tres de los casos se incluyeron en más de una categoría, por presentar ecográficamente dos anomalías esqueléticas no asociadas nosológicamente a priori. Un 71,4% de los casos (40/56) correspondió a anomalías del esqueleto radial (figs. 1–3) y un 8,9% (5/56), a anomalías del esqueleto axial.
Anomalías esqueléticas diagnosticadas en la Sección de Medicina Fetal del Hospital General Universitario Gregorio Marañón de acuerdo con la clasificación de Swanson et al2.
| Tipo | n (%) | Presentación | |
| Aislada, n (%) | Malformaciones asociadas, n (%) | ||
| Anomalías focales del esqueleto radial | 40/56 (71,4) | 17/40 (42,5) | 23/40 (57,5) |
| Falta de formación de una parte | 3/40 (7,5) | 1 | 2 |
| Defectos transversos terminales | 1 | 1 (Aqueiria) | - |
| Ectrodactilia | 2 | - | 2 |
| Falta de diferenciación y anomalías posicionales | 27/40 (67,5) | 11 | 16 |
| Contracturas generalizadas | 1 | 1 (Artrogriposis) | - |
| Defectos posicionales del pie | 14 | 8 | 6 |
| Pie equinovaro unilateral | 9 | 5 | 4 |
| Pie equinovaro bilateral | 5 | 3 | 2 |
| Defectos posicionales de la mano | 3 | - | 3 |
| Mano en garra | 3 | - | 3 |
| Sindactilia | 1 | 1 | - |
| Clinodactilia | 4 | 1 | 3 |
| Pies y manos mal posicionados (sin especificar) | 4 | - | 4 |
| Duplicación | 2/40 (5,0) | 1 | 1 |
| Polidactilia | 2 | 1 | 1 |
| Subcrecimiento | 8 (20,0) | 5 | 3 |
| Huesos largos cortos (sin especificar) | 8 | 5 | 3 |
| Banda amniótica constrictiva (Pie equinovaro bilateral) | 1/40 (2,5) | - | 1 |
| Anomalías del esqueleto axial | 5/56 (8,9) | 3/5 (60,0) | 2/5(40,0) |
| Teratoma sacrococcígeo | 2 | 2 | - |
| Escoliosis y cifosis | 3 | 1 | 2 |
| Estructurales | 2 | 1 | 1 |
| Posicionales | 1 | - | 1 |
| Osteocondrodisplasias | 14/56 (25,0) | 6/6 (100,0) | 0 |
| Displasias esqueléticas letales | 6/14 (42,9) | 6 | 0 |
| Displasia tanatofórica | 4 | 4 | 0 |
| Acondrogénesis | 2 | 2 | 0 |
| Displasias esqueléticas no letales o de letalitat variable | 3/14 (21,4) | 3 | 0 |
| Acondroplasia heterocigota | 1 | 1 | 0 |
| Osteogénesis imperfecta | 2 | 2 | 0 |
| Displasias sin especificar | 6/14 (42,9) | 6 | 0 |



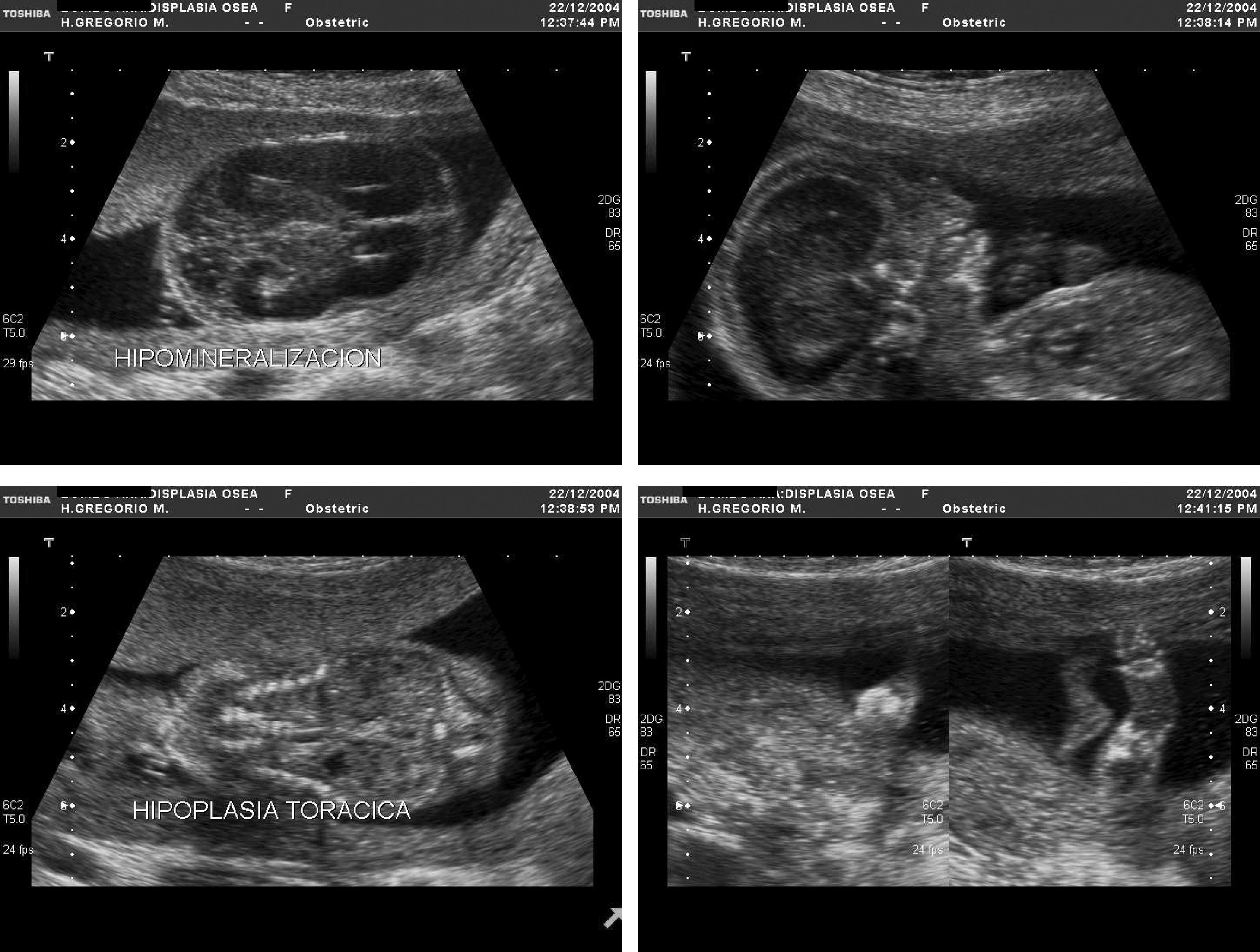
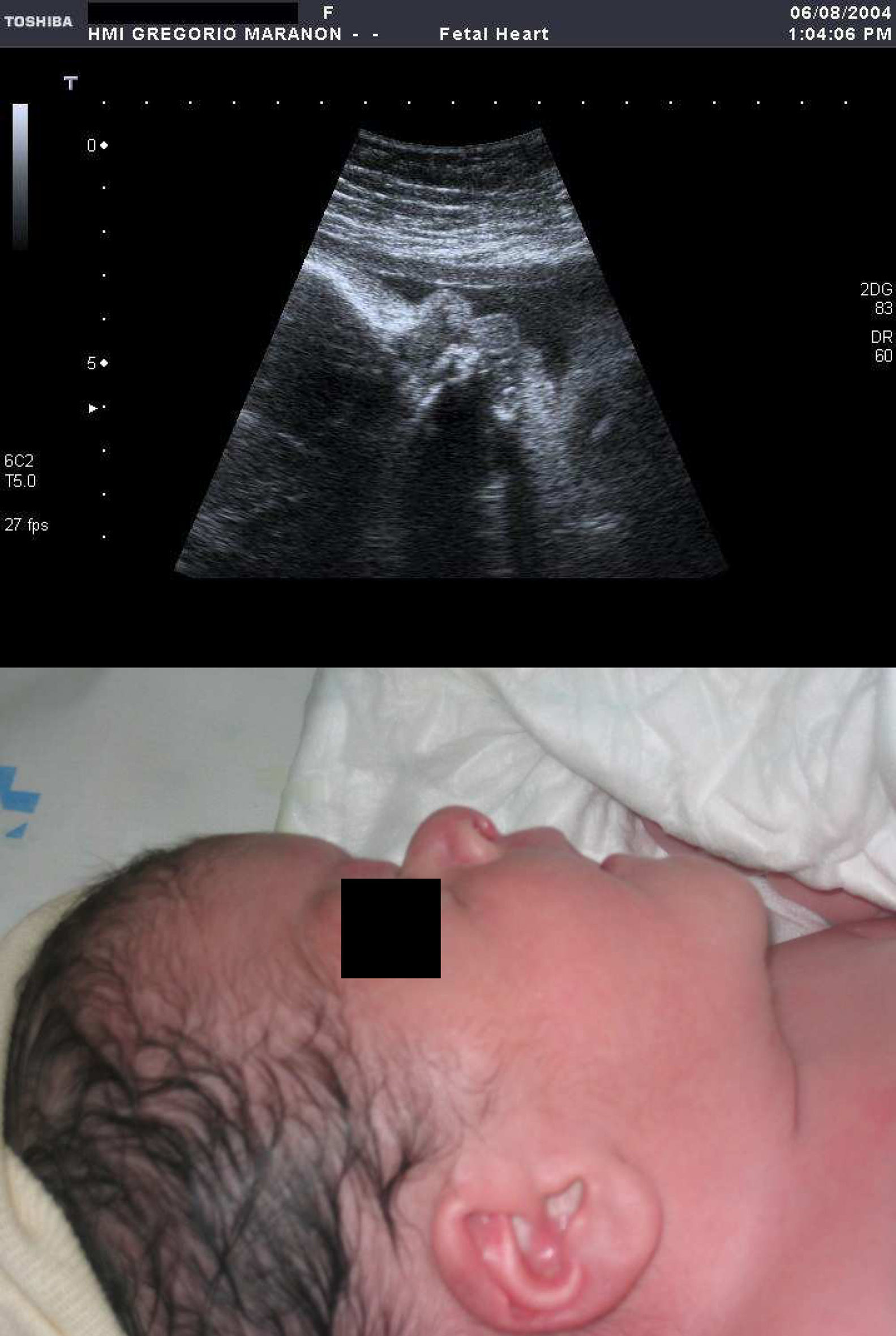
Respecto a las osteocondrodisplasias, se estableció el diagnóstico ecográfico de sospecha en 14 casos (14/56; 25%), 6 de ellas letales (4 casos de displasia tanatofórica y 2 de acondrogénesis) (fig. 4), 3 no letales (2 casos de osteogénesis imperfecta (fig. 5) y 1 de acondroplasia heterocigota (fig. 6) y 5 casos de displasias no etiquetadas.
, abombamiento frontal (4b), hipoplasia torácica (4c), micromelia (4d).")

; perfil del recién nacido (6b).")
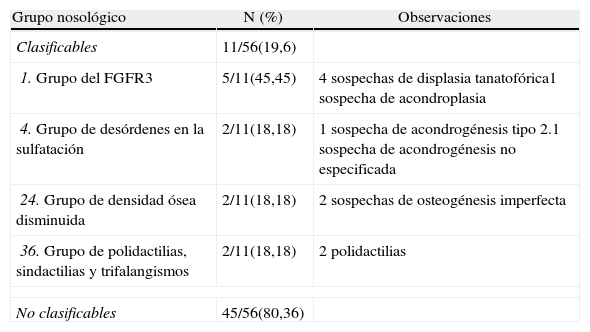
Atendiendo a la etiología genética, sólo pudieron clasificarse 11 casos (19,6%) (tabla 2)5.
Resultados según la clasificación del Nosology Group of the International Skeletal Dysplasia Society5
| Grupo nosológico | N (%) | Observaciones |
| Clasificables | 11/56(19,6) | |
| 1. Grupo del FGFR3 | 5/11(45,45) | 4 sospechas de displasia tanatofórica1 sospecha de acondroplasia |
| 4. Grupo de desórdenes en la sulfatación | 2/11(18,18) | 1 sospecha de acondrogénesis tipo 2.1 sospecha de acondrogénesis no especificada |
| 24. Grupo de densidad ósea disminuida | 2/11(18,18) | 2 sospechas de osteogénesis imperfecta |
| 36. Grupo de polidactilias, sindactilias y trifalangismos | 2/11(18,18) | 2 polidactilias |
| No clasificables | 45/56(80,36) | |
De las 54 gestaciones en las que se pudo realizar un seguimiento completo, la tasa de ILE fue del 29,6% (16/54) y en un caso se produjo un fallecimiento intraútero a las 23 semanas en un feto afectado de una displasia esquelética con criterios ecográficos de letalidad. En un 68,5% (37/54) la gestación siguió su curso. Se registró una muerte intraparto en un cuadro polimalformativo a las 31 semanas con polihidramnios grave que acudió al hospital en trabajo de parto. Hubo tres muertes neonatales tempranas, secundarias a una prematuridad extrema (dos partos en las semanas 26 y 27), y a la propia afección fetal (teratoma sacrococcígeo). En un 61,1% (33/54) la gestación finalizó con la obtención de un recién nacido vivo que superó el período neonatal.
La EG media al parto fue de 36 semanas (rango 26-41; DE 5,24). En un 24,3% (9/37) la EG fue inferior a la semana 37; en un 13,5% (5/37), inferior a la semana 34, y en un 10,8% (4/37), inferior a la 32. La tasa de cesáreas alcanzó el 35,1% (13/37). El peso medio al nacer del recién nacido fue de 2.548g (rango 717-3.970; DE 883) y el test de Apgar a los 5 minutos inferior a 7 en el 10,8% (4/37).
DiscusiónEn el presente estudio se han analizado los datos de una serie de 56 casos de anomalías esqueléticas diagnosticadas mediante ecografía en nuestro centro. Del total de estas anomalías, un 46,4% se presentó de forma aislada; un 25%, asociada a otras malformaciones, y un 28,6%, en el contexto de un cuadro polimalformativo. El 61,1% de estas gestaciones finalizó con la obtención de un recién nacido vivo; un 29,6% optó por una ILE, y en un 9,3% se produjo fallecimiento intrauterino o muerte perinatal.
Las anomalías esqueléticas son un tipo de malformaciones infrecuentes con una incidencia perinatal en torno a 1/2.000-4.000 nacimientos6. En nuestra casuística, de un total de 45.630 partos atendidos en nuestro centro durante el período de estudio, la incidencia fue de 1/815 (12,27 casos/10.000 partos). Esta incidencia es superior a la recogida en la bibliografía, probablemente porque nuestro hospital es centro de referencia de afección fetal.
El diagnóstico prenatal ecográfico de las displasias esqueléticas letales se suele establecer alrededor de las 18-20 semanas de gestación, coincidiendo con la realización de la ecografía morfológica del segundo trimestre7. Especialmente en las formas menos graves, hay un segundo pico diagnóstico en torno a la semana 30 de gestación, relacionado con la aparición de complicaciones asociadas a algunas de las displasias, como el polihidramnios8. En nuestra muestra se ha comprobado esta misma tendencia, con dos picos de EG al diagnóstico: el primero, coincidiendo con la semana 20 de gestación, y, el segundo, en la semana 35. Se ha considerado como momento del diagnóstico aquél en el que la paciente fue valorada por primera vez en la Sección de Medicina Fetal de nuestro centro. Al ser un hospital terciario, el segundo pico de diagnóstico tardío en la semana 35 podría explicarse por el elevado número de derivaciones para una segunda opinión, completar estudio o para control y finalización de la gestación. No obstante, a pesar de esta distribución, el 55,4% (31/56) de las anomalías se diagnosticó (o nos remitieron a la paciente) antes de la semana 22 de gestación.
En nuestra serie, en el 51,8% (29/56) de los casos se realizó una prueba invasiva para estudio citogenético, y en el 34,5% de ellas (10/29) se diagnosticó una anomalía cromosómica, la mitad trisomías 21. La incidencia de cromosomopatía alcanzó el 58,3% (7/12) cuando la alteración ósea se presentó en el contexto de un cuadro polimalformativo. Hasta en un 25% (14/56) del total de los casos se diagnosticó otra malformación asociada.
La mayor parte de los diagnósticos fueron hallazgos focales (45/56; 80,4%) con afectación de extremidades (40/56; 71,4%) o columna vertebral (5/56; 8,9%). Este tipo de hallazgos ecográficos son los más comunes y la repercusión de ellos en el neonato suele ser limitada, siempre que se presenten de forma aislada. Entre estas anomalías, las detectadas con más frecuencia son las que afectan a la posición de las extremidades, especialmente de manos y pies (17/56; 37,5%) y al número y posición de los dedos (polidactilias, clinodactilias, sindactilias) (7/56; 12,5%).
Entre los diagnósticos de sospecha de osteocondrodisplasia predominaron las letales, como la displasia tanatofórica, sobre las anomalías no letales, como la acondroplasia y algunos tipos de osteogénesis imperfecta. Sin embargo, es preciso resaltar que hasta en un 42,9% (6/14) de los casos no pudo establecerse un diagnóstico específico según las clasificaciones más recientes2,9,10. Estos datos están en consonancia con la dificultad recogida en la bibliografía para catalogar este tipo de malformaciones basándose exclusivamente en los hallazgos ecográficos11.
Es preciso señalar que aunque la ecografía es una herramienta diagnóstica muy valiosa, su principal desventaja radica en la dificultad para establecer un diagnóstico etiológico preciso7,11. Con esta premisa, la mayoría de los autores defienden que ante la dificultad de proporcionar a los padres un diagnóstico exacto, puede ser de mayor utilidad orientar la displasia de acuerdo con la letalidad12. Para ello se dispone de una serie de criterios indicativos de letalidad, como son la micromelia grave, la hipoplasia torácica grave o rasgos específicos de una displasia letal particular.
En la mayoría de los casos, la osteocondrodisplasia aparece como una alteración esporádica, no precedida de antecedentes familiares de otros trastornos esqueléticos. La etiología habitual suele ser una alteración monogénica, resultado de diferentes mutaciones con expresión y herencia variables. De la serie presentada, únicamente en uno de los casos de sospecha de displasia ósea se pudo diagnosticar la mutación causante y, en virtud de ella, el diagnóstico de presunción. Parece que el desarrollo y la paulatina incorporación de las nuevas técnicas genéticas (microarrays, FISH) podrían suponer una herramienta fundamental a la hora de establecer el diagnóstico y ubicar la afección fetal de acuerdo con la clasificación propuesta por la ISDS5. No obstante, conviene recordar que una misma mutación puede ser origen de un amplio espectro de displasias esqueléticas de diferente pronóstico1.
Cabe destacar que menos del 20% de los casos fueron clasificables de acuerdo con las últimas directrices de la ISDS. Esto se debe al énfasis que esta clasificación pone en el origen genético de la anomalía, el cual es difícil de establecer en un porcentaje elevado de casos. Por el momento, parece más práctico emplear otras clasificaciones en nuestra práctica clínica habitual, como las guías EUROCAT10, creadas para proporcionar a los facultativos no especializados en dismorfología herramientas para el tratamiento de malformaciones congénitas.
Los resultados gestacionales en nuestra muestra son reflejo de la gravedad de la afección estudiada. En un 29,6% (16/54) de los casos los padres solicitaron una ILE. La mayoría se llevaron a cabo ante la sospecha de una displasia esquelética letal o un cuadro polimalformativo. Todas ellas se realizaron en centros externos, por lo que no disponemos de resultados anatomopatológicos. Un 9,3% (5/54) resultó en muerte fetal intrauterina o neonatal temprana.
Respecto a la vía del parto, sólo se planteará la cesárea electiva, además de por otras indicaciones obstétricas, cuando haya riesgo de complicaciones en relación con la propia afección fetal (fracturas en osteogénesis imperfecta, macrocefalia, artrogriposis). En nuestra serie, de las 37 gestaciones que finalizaron con un parto, el 64,9% (24/37) se asistió por vía vaginal. La alta tasa de prematuridad en estas gestaciones (que se halla próxima al 15% por debajo de las 34 semanas en nuestra serie) parece estar en relación con la presencia de polihidramnios, crecimiento intrauterino restringido y otras malformaciones fetales.
Las anomalías esqueléticas son un defecto del desarrollo fetal intrauterino infrecuente. La repercusión de éste depende en gran medida de su gravedad, y en este sentido las osteocondrodisplasias son de especial relevancia dada su elevada morbimortalidad. La evaluación ecográfica fetal facilita el diagnóstico prenatal de estas alteraciones; sin embargo, presenta limitaciones, especialmente en lo que se refiere a establecer un diagnóstico específico. El estudio de las displasias esqueléticas debe incorporar la realización de un estudio genético que permita el diagnóstico etiológico y el asesoramiento prenatal. El diagnóstico debe confirmarse en el recién nacido y, en caso de ILE, fallecimiento intraútero o neonatal, realizar un correcto estudio radiológico y anatomopatológico.
Conflicto de interesesLos autores declaran la inexistencia de implicaciones comerciales en el presente trabajo.
Datos preliminares del presente trabajo han sido presentados en forma de póster en XXVI Congreso Nacional de Ecografía Obstétrico-Ginecológica (SESEGO); Marbella, 10 de julio de 2010.