


a Síndrome do Coração Esquerdo Hipoplásico é uma cardiopatia complexa, caracterizada pelo subdesenvolvimento das estruturas cardíacas esquerdas. Pode ser diagnosticada a partir das 18 semanas de gestação através da ecocardiografia fetal transabdominal.
Objectivodescrever a abordagem de uma equipa de Diagnóstico Pré-natal de um centro de referência materno-infantil no contexto da Síndrome do Coração Esquerdo Hipoplásico, entre Janeiro de 1990 e Dezembro de 2008 e o seguimento dos casos diagnosticados.
Material e métodosanálise retrospectiva dos casos com o diagnóstico pré-natal de Síndrome do Coração Esquerdo Hipoplásico nos últimos 19 anos. Foram analisados os seguintes parâmetros: ano de diagnóstico, motivo de referência à consulta de cardiologia fetal, idade gestacional do diagnóstico, idade gestacional da interrupção médica da gravidez, mortes fetais, dados da necrópsia, antecedentes familiares de cardiopatia, peso de nascimento, idade gestacional dos recém-nascidos e respectivo seguimento.
Resultadosdurante o período em análise foram diagnosticadas na Consulta de Cardiologia Fetal 311 cardiopatias congénitas e destas 67 (21.5%) correspondiam a Síndrome do Coração Esquerdo Hipoplásico. Vinte e nove (43.3%) casais optaram pela interrupção médica da gravidez. A idade gestacional mediana de interrupção médica da gravidez foi de 24 semanas, variando de 20 a 35 semanas. Nestes casos, a necrópsia foi concordante com o diagnóstico de Síndrome do Coração Esquerdo Hipoplásico. Registaram-se quatro mortes fetais. Dos 34 recém-nascidos vivos, oito faleceram no período neonatal precoce. Dos 12 recém-nascidos sujeitos a cirurgia cardíaca, seis estão vivos, três dos quais após dois anos de conclusão do terceiro estadio da cirurgia paliativa de Norwood.
ConclusõesApesar da possibilidade de um diagnóstico precoce, do avanço das técnicas percutâneas, cirúrgicas e cuidados intensivos neonatais, trata-se de uma patologia com uma morbimortalidade significativa associada, pelo que a sua abordagem continua a ser um desafio.
the Hypoplastic Left Heart Syndrome is due to the underdevelopment of left-sided cardiac structures. This syndrome can be diagnosed from 18 weeks of gestation through transabdominal fetal echocardiography.
Aimsdescription of a prenatal diagnosis team approach in the context of Hypoplastic Left Heart Syndrome, from January 1990 to December 2008, and case follow-up.
Material and methodsretrospective analysis of the Hypoplastic Left Heart Syndrome cases diagnosed prenatally during the course of 19 years. The following parameters were analysed: year of diagnosis, reason for referral, gestational age at diagnosis, gestational age of medical termination of the pregnancy, necropsy findings, fetal deaths, family past history of congenital heart disease, birth weight, gestational age of the newborns and follow-up.
Resultsduring the studied period 311 congenital heart disease were diagnosed in our department, 67 (21.5%) of which with Hypoplastic Left Heart Syndrome. Twenty-nine (43.3%) parents opted for medical termination of the pregnancy. The median gestational age for medical abortion was 24 weeks, varying from 20 to 35 weeks. In these cases the necropsy confirmed the prenatal diagnosis of Hypoplastic Left Heart Syndrome. There were four fetal deaths. Of the 34 newborns, eight died in the early neonatal period. Regarding the 12 newborns that underwent surgical correction, six are alive; three of them two years post the completion of the Norwood's third stage.
Conclusiondespite advances in neonatal intensive care, surgical techniques and percutaneous therapy, the morbidity and mortality associated with this pathology raises several ethical issues.
A Síndrome do Coração Esquerdo Hipoplásico (SCEH) representa 1-3.8% do total de cardiopatias congénitas. Apresenta uma incidência de aproximadamente 0.016-0.036% dos nados-vivos, sendo responsável por 23% das mortes causadas por cardiopatia no período neonatal. Define-se como o desenvolvimento anormal das estruturas cardíacas esquerdas, condicionando o débito sistémico. Engloba o subdesenvolvimento do ventrículo esquerdo, aorta e arco aórtico, bem como atrésia ou estenose da válvula mitral1–3. Estudos epidemiológicos reportam influências multifactoriais em 90% dos casos. As anomalias cromossómicas são responsáveis por cerca de 6% de todas as cardiopatias congénitas1. Nos últimos 50 anos, têm aumentado os relatos que suportam uma etiologia genética4–6. Foi identificado um risco de recorrência em irmãos de 0.5% e uma taxa de recorrência em gravidezes posteriores de 2 a 6%1,5,6. O exame complementar de diagnóstico de eleição é a ecocardiografia bidimensional com estudo Doppler. Esta Síndrome pode ser diagnosticada a partir das 18 semanas de gestação, através da ecocardiografia fetal.
Após o diagnóstico, deve ser feita uma avaliação adicional incluindo a avaliação genética e a exclusão de outras malformações extracardíacas, uma vez que 25% dos fetos afectados têm uma doença genética associada ou uma anomalia major extracardíaca1. A maioria dos recém-nascidos (RN) afectados é de termo, com peso adequado à idade gestacional1,2. O grau de obstrução ao fluxo sanguíneo sistémico determina a precocidade e gravidade das manifestações clínicas3. Com os progressos da medicina intensiva e das técnicas cirúrgicas e percutâneas pré e pós-natais, incluindo o recurso à transplantação cardíaca, a mortalidade precoce tem vindo a decrescer1,2.
Foi nosso objectivo descrever a abordagem multidisciplinar de uma equipa de diagnóstico pré-natal no contexto da SCEH, nos últimos 19 anos.
Material e métodosFoi efectuada uma análise retrospectiva dos processos clínicos das grávidas e crianças com o diagnóstico pré-natal de SCEH, entre Janeiro de 1990 e Dezembro de 2008. Foram avaliados os seguintes parâmetros: ano de diagnóstico, motivo de referência à consulta de cardiologia fetal, idade gestacional do diagnóstico, idade gestacional da interrupção médica da gravidez (IMG), mortes fetais, dados da necrópsia, antecedentes familiares de cardiopatia, peso de nascimento, idade gestacional dos recém-nascidos e respectivo seguimento.
A todos os casais foi facultada informação relativa à natureza e implicações desta cardiopatia, as possíveis hipóteses terapêuticas existentes no nosso Centro, assim como a natureza paliativa da correcção cirúrgica usada nestes casos (cirurgia de Norwood) e os respectivos resultados. Foi recomendado a todas as grávidas a realização de amniocentese para estudo de cariótipo fetal, incluindo a exclusão da delecção 22q11. Em caso de IMG, era efectuada necrópsia ao feto. O seguimento foi realizado num centro de referência com apoio de um Serviço de Cirurgia Cardiotorácica com experiência pediátrica para planeamento atempado da intervenção. Todos os casais foram referenciados à Consulta de Aconselhamento Genético.
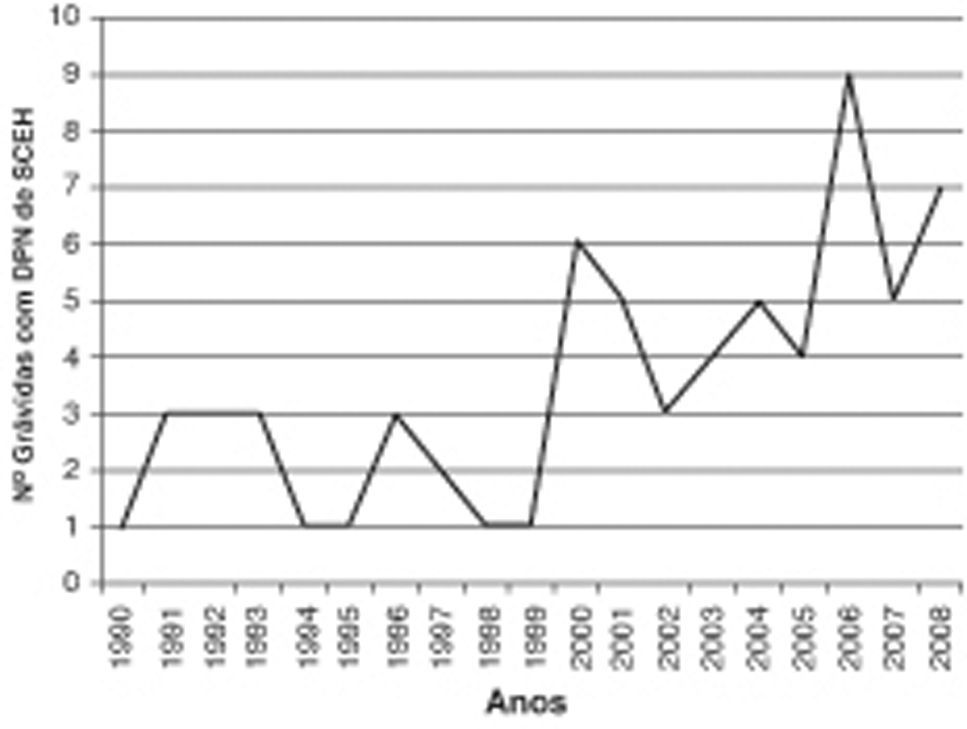
ResultadosNos últimos 19 anos, foram diagnosticadas 311 cardiopatias congénitas, e destas, 67 (21.5%) correspondiam a SCEH. A distribuição anual não tem sido regular, tendo-se desde 2000 assistido a um aumento do número de grávidas referenciadas com esta hipótese diagnóstica, registando-se 48 casos (71.6% dos diagnósticos efectuados) (fig. 1).
.")
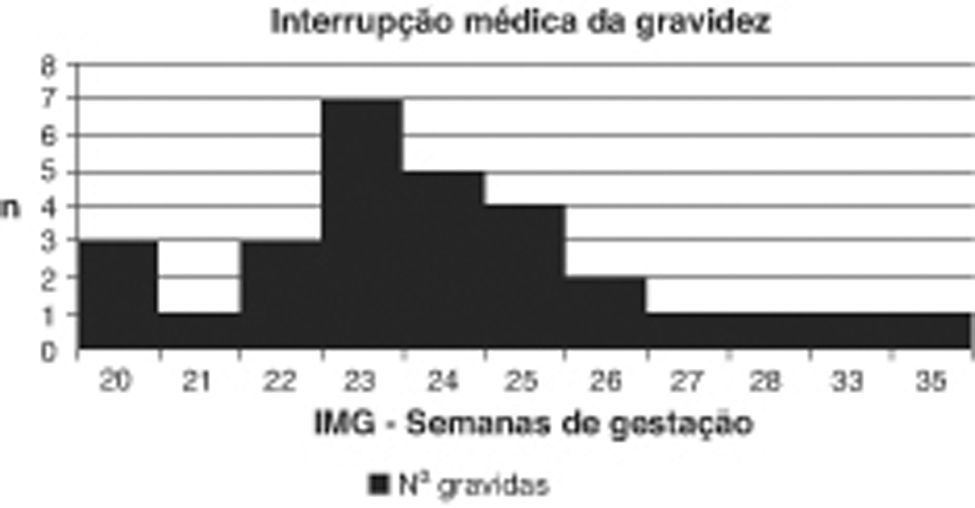
A idade gestacional média de diagnóstico foi de 22±3.3 semanas. Vinte e nove (43.3%) casais optaram pela IMG. A mediana da idade gestacional de IMG foi de 24 semanas (20 a 35 semanas) (fig. 2). Nestes casos, a necrópsia confirmou em todos os fetos hipoplasia do ventrículo esquerdo. Vinte (69%) tinham associado hipoplasia da aorta ascendente e/ou do arco aórtico e nove (31%) coarctação aórtica severa. Observou-se fibroelastose endocárdica em oito fetos (27.6%). Quanto a anomalias da válvula mitral, um (3.4%) apresentava atrésia da válvula mitral e 28 (96.6%) estenose da válvula mitral. Em seis fetos (20.7%) observou-se foramen ovale restritivo.
.")
Foram realizadas 63 amniocenteses para estudo do cariótipo fetal, tendo-se identificado uma trissomia 18 (restrição do crescimento intra-uterino, fenda palatina e rins em ferradura) e uma delecção da região q11.2 do cromossoma 22 (fenda palatina, dismorfismos faciais, nomeadamente fendas palpebrais estreitas e microcefalia). Para além destes casos, três fetos apresentavam síndromes polimalformativas (um com hipoplasia das glândulas supra-renais e hidrâmnios; outro com restrição do crescimento intra-uterino e anencefalia e o terceiro caso com restrição do crescimento intra-uterino e ossos longos curtos). Todos estes casos foram submetidos a IMG. Dois casais tinham filho anterior com o diagnóstico de SCEH, correspondendo a 3% da amostra e uma grávida tinha uma cardiopatia congénita simples (Comunicação Interauricular operada).
Registaram-se quatro mortes fetais (6% dos fetos). A incidência da SCEH, com diagnóstico pré-natal, foi de 0.05% do total de nados-vivos. Dos 34 RN vivos, 21 eram do sexo masculino (61.8%). A maioria era de termo (32; 94.1%). Dois dos RN eram prematuros, com idade gestacional de 32 semanas e todos apresentavam peso de nascimento adequado à idade gestacional. Após o nascimento foram transferidos para a Unidade de Cuidados Intensivos Neonatais, para monitorização e terapêutica endovenosa com Prostaglandina E1, sendo posteriormente referenciados ao Serviço de Cirurgia Cardiotorácica de referência. Oito faleceram no período neonatal precoce por insuficiência cardíaca refractária, sendo um dos casos prematuro. Neste grupo, não se identificaram outros processos patológicos que conduzissem à morte.
Doze RN foram submetidos a cirurgia cardíaca, tendo seis concluído o terceiro estadio da cirurgia paliativa de Norwood. Quatro crianças faleceram após o primeiro estadio e duas após o segundo estadio, tendo uma sido submetida à implantação de um conducto entre o ventrículo direito e a artéria pulmonar (procedimento de Sano) no primeiro estadio. Seis das crianças operadas estão vivas, três das quais após dois anos de conclusão do terceiro estadio. Uma destas crianças, actualmente com cinco anos, apresenta atraso de desenvolvimento psicomotor, microcefalia e episódios de Acidente Isquémico Transitório Cerebral. Registaram-se múltiplos internamentos por insuficiência cardíaca descompensada, sobretudo após o primeiro estadio da cirurgia paliativa de Norwood. A mortalidade global dos nados-vivos, incluindo os casos que foram submetidos aos vários estadios da cirurgia de Norwood ou modificações desta, foi de 82.4%.
DiscussãoAssistimos, ao longo dos anos, a um aumento do número de casos de diagnóstico pré-natal de SCEH, face ao melhor conhecimento desta patologia por parte dos clínicos e aos avanços tecnológicos a nível da ecocardiografia fetal. Esta síndrome pode ser diagnosticada entre as 18 e as 22 semanas de gestação1. Perante o diagnóstico, é imprescindível facultar aos casais a informação relativa às implicações subjacentes a esta patologia e opções terapêuticas existentes. Esta informação provavelmente condicionou a opção da IMG em cerca de metade dos casais. Constataram-se anomalias cromossómicas em dois casos, correspondendo a 3% da amostra, inferior ao descrito em outros trabalhos publicados1,2. Este achado deve-se provavelmente ao facto dos casos com anomalias cromossómicas serem submetidos a IMG nos centros de origem. A necrópsia confirmou o diagnóstico em todos os casos e em alguns acrescentou dados que condicionam o prognóstico da doença, nomeadamente a existência de fibroelastose endocárdica e a presença de foramen ovale restritivo em oito e seis fetos, respectivamente.
Dois dos casais tinham filho anterior com SCEH (3% da amostra), o que demonstra a forte carga genética, como relatado na literatura1,4–7. A incidência de SCEH no nosso estudo (0,05%) foi ligeiramente superior à enumerada na literatura (0.016-0.036%)1,2. Esta incidência claramente reflecte o facto do estudo ser realizado num Centro de Referência, não se podendo reportar à população em geral.
A grande maioria dos RN era de termo, com peso adequado à idade gestacional, tal como referido na literatura científica1,2. À semelhança de outras séries publicadas1,2, a doença atingiu predominantemente o sexo masculino (61.8%) e verificou-se uma elevada mortalidade no período neonatal.
Todos os casos de SCEH foram submetidos ao procedimento de Norwood. A taxa de sobrevivência desta doença é descrita na literatura médica como sendo de cerca de 50-60% nos primeiros dois anos após o terceiro estadio8–12. Na nossa revisão a sobrevivência dos casos operados é actualmente de 50%. Em 2003, Sano et al13 publicaram os resultados da modificação do primeiro estadio, nomeadamente, substituindo o shunt Blalock-Taussig modificado por um conducto entre o ventrículo direito e a artéria pulmonar, como fonte de fluxo pulmonar (procedimento de Sano). Têm sido realizados vários estudos comparativos entre as duas técnicas10–13. Estes estudos referem uma menor mortalidade entre os estadios e maior sobrevida a médio prazo com o procedimento de Sano (variando de 70 a 80%)10–13. Outra modalidade terapêutica é o recurso à transplantação cardíaca. Existem trabalhos publicados2,14 que referem que esta última se associa a uma maior mortalidade, pelo tempo de espera associado. O tratamento híbrido, combinando a abordagem cirúrgica e percutânea, tem vindo a apresentar resultados promissores15–17. O cateterismo de intervenção fetal, quer através da atriosseptostomia ou valvuloplastia aórtica, a fim de manter o desenvolvimento do ventrículo esquerdo é outra opção terapêutica viável, parecendo associar-se a uma melhoria da sobrevida18,19. A intervenção fetal apesar de já ter sido realizada no nosso Centro no contexto de outras patologias, na SCEH não foi realizada até à data, por falta de consentimento dos pais.
ConclusõesA SCEH permanece uma das mais desafiantes e menos compreendidas formas de doença cardíaca congénita. Com os avanços tecnológicos, do aperfeiçoamento das técnicas cirúrgicas e das percutâneas, a mortalidade a curto prazo da SCEH melhorou significativamente. No entanto, a sua abordagem médico-cirúrgica mantém-se complexa. Questões pertinentes permanecem no que diz respeito ao tratamento e respectivos resultados funcionais e cognitivos a longo prazo. O diagnóstico pré-natal é imprescindível para um tratamento médico e cirúrgico atempado, permitindo, a curto prazo, a diminuição da morbilidade e mortalidade associadas a esta patologia.