


Presentamos un caso de diagnóstico prenatal de un feto afecto de trisomía parcial del cromosoma 6 y monosomía parcial del cromosoma X:46X der(X)t(X;6)(p22.3;21.1). Dicho desbalance proviene de una translocación balanceada detectada en el cariotipo materno: 46XX t(X:6)(p22.3;p21.1). El diagnóstico se hizo mediante cariotipo fetal en líquido amniótico tras detectar un retraso severo del crecimiento fetal (menor de un percentil 3), tras descartar otras causas como origen del retraso. El neonato presenta una facies dismórfica, acompañada de dificultades para la alimentación y retraso del crecimiento y psicomotor, descritos en otros casos de trisomía parcial del cromosoma 6. Además presenta microftalmia y esclerocórnea asociadas a la microdeleción del cromosoma X en la región Xp22.3. Estos signos característicos nos permitieron hacer el diagnóstico clínico de síndrome de MIDAS.
We present a prenatal diagnosis of a foetus affected by partial trisomy 6 p and partial monosomy X, 46X der(X)t(X;6)(p22.3;21.1), resulting from a maternal balanced translocation: 46XX t(X:6)(p22.3;p21.1) The diagnosis was made by cytogenetic analysis of amniotic fluid, after intrauterine growth retardation was detected (<3rd centile). Other causes were investigated. The newborn has a dysmorphic face, feeding difficulties, and growth and mental retardation. These characteristic features appear to make partial trisomy 6 p a clinically recognisable syndrome. Microphthalmia and sclerocornea were also present, both associated with microdeletion of Xp22.3 region. These characteristic signs led to the clinical diagnosis of MIDAS syndrome.
Ante el diagnóstico de un retraso de crecimiento intrauterino (peso fetal estimado menor de un percentil 10) debe hacerse un estudio detallado que incluya estudio morfológico fetal, doppler de arteria umbilical, arteria cerebral media y arterias uterinas, despistaje de preeclampsia, serología TORCH, y lúes y cariotipo fetal.
En el caso que presentamos los resultados de las pruebas anteriores fueron normales excepto un peso fetal estimado (PFE) menor del percentil 3 y un cariotipo fetal alterado, con una trisomía parcial del cromosoma 6 y una monosomía parcial del cromosoma X: 46X der(X)t(X;6)(p22.3;21.1). Dicha translocación parece proceder de un desbalance heredado de la madre, portadora sana de una translocación recíproca entre los cromosomas X y 6, con puntos de corte en la banda p22.3 del cromosoma X y en la banda p21.1 del cromosoma 6.
Los casos descritos en la literatura de trisomía parcial 6p21.1 a 6pter mostraron: retraso del crecimiento pre o posnatal, una amplia variedad de anomalías menores y algunas malformaciones mayores características como microcefalia, craneosinostosis, hidrocefalia, alteraciones renales y malformaciones cardiacas entre otras1–3. Estos pacientes son propensos a las infecciones4. Los casos de deleción parcial Xp22.3 a Xpter encontrados en la bibliografía mostraron microftalmia, lesiones dérmicas, agenesia del cuerpo calloso y malformaciones cardiacas como defectos más frecuentes5,6.
La paciente presentó al nacimiento una facies dismórfica, acompañada de dificultades para la alimentación y retraso del crecimiento. Las alteraciones oculares (microftalmia, esclerocórnea) y la microdeleción de Xp22.3 nos ayudaron al diagnóstico de síndrome de MIDAS o MLS7.
Caso clínicoPaciente secundigesta, sin antecedentes de interés, con gestación normoevolutiva hasta la realización de la ecografía morfológica en la semana 20, en la que se detecta un retraso del crecimiento fetal (3 semanas menor que amenorrea). En la primera ecografía la longitud craneocaudal correspondía con una semana menos que amenorrea, por lo que se modificó la fecha de última regla por ecografía, y el decalaje en el momento de realizar la ecografía morfológica era de 2 semanas. La paciente rechazó la realización de una amniocentesis genética, siguiendo controles cada 2 semanas. Se le realizó una serología TORCH, parvovirus B19 y lúes, siendo estas negativas, anticuerpos anticardiolipina negativos y estudio doppler dentro de la normalidad. Los controles analíticos y de tensión arterial eran normales, sin proteinuria.
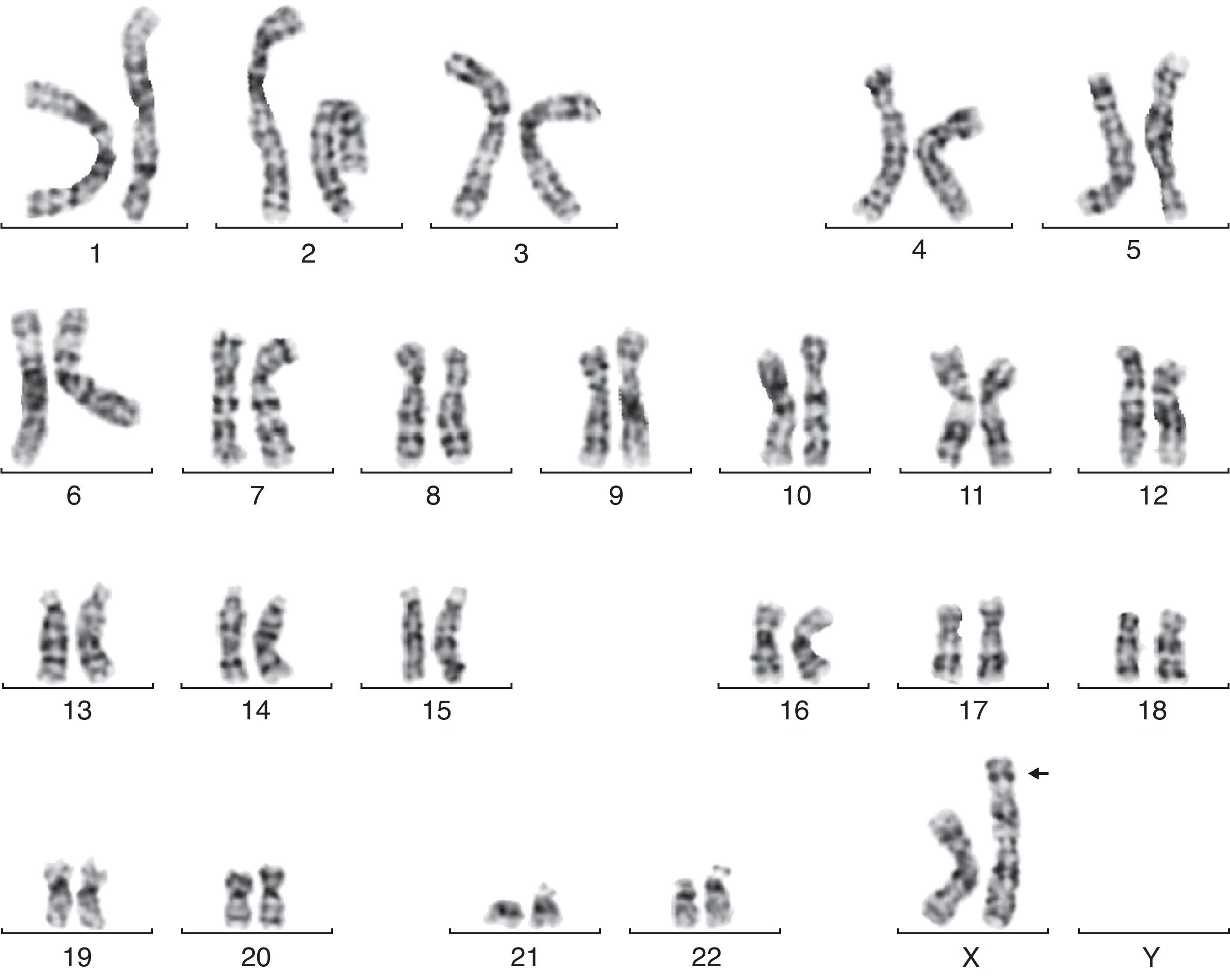
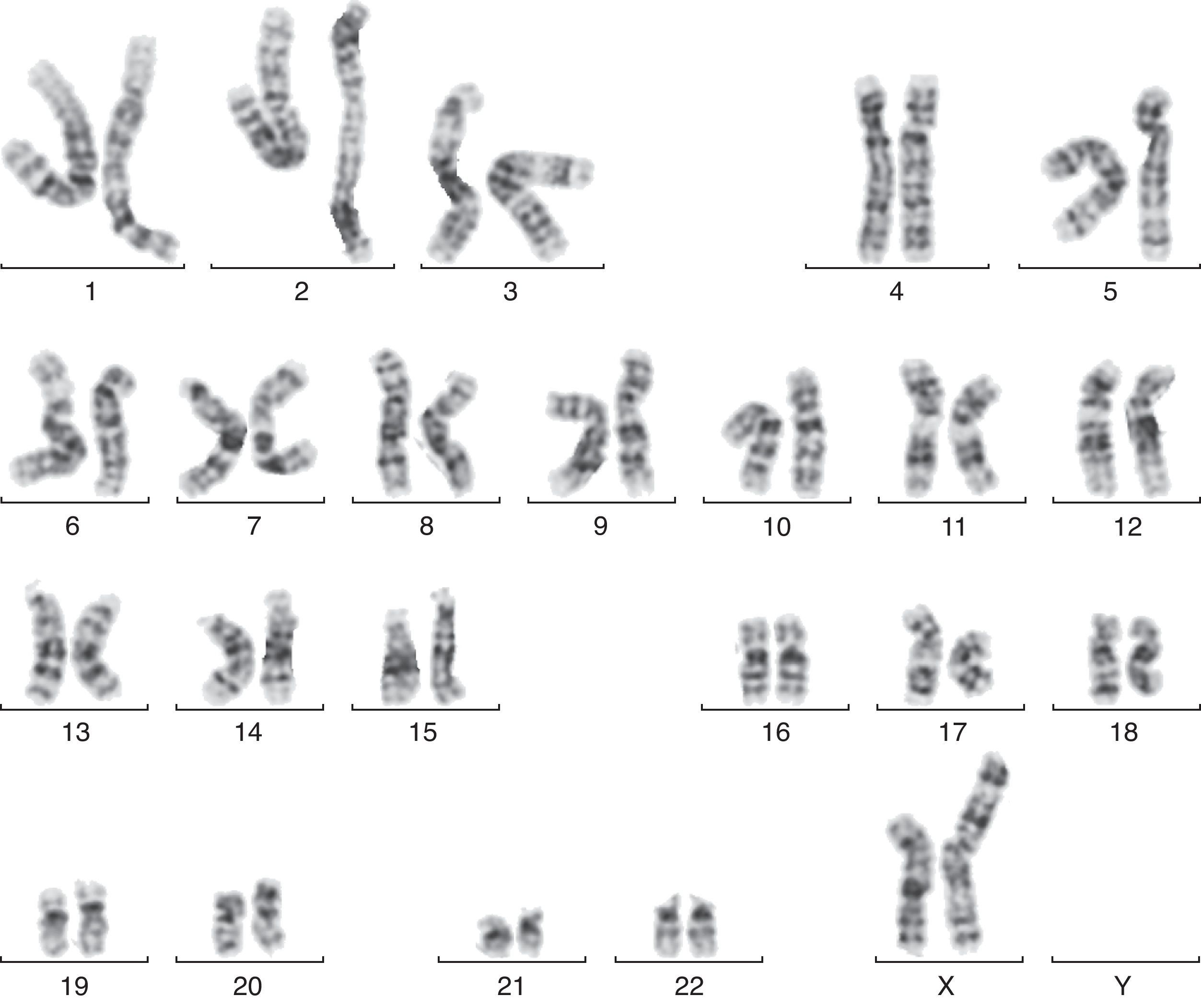
En la semana 26, tras confirmar que el PFE era menor del percentil 3, se ofrece de nuevo la realización de una amniocentesis genética y la paciente acepta. Se lleva a cabo una técnica FISH que es informada como feto XX normal, pero el cariotipo definitivo detecta una trisomía parcial del cromosoma 6 y una monosomía parcial del cromosoma X: 46X der(X)t(X;6)(p22.3;21.1) (fig. 1). Se realiza cariotipo a los padres en sangre periférica, siendo la madre portadora de de una translocación recíproca entre los cromosomas X y 6, con puntos de corte en la banda p22.3 del cromosoma X y en la banda p21.1 del cromosoma 6 (fig. 2). Dicha translocación es aparentemente balanceada, por lo que la paciente es portadora sana, pero tiene riego de formar gametos cromosómicamente desbalanceados, como ha ocurrido en el caso de este feto. Se recomienda diagnóstico prenatal o preimplantacional en futuras gestaciones, así como el estudio de cariotipo en los familiares de primer grado del paciente para detectar otros posibles portadores de dicha translocación.
t(X;6)(p22.3;21.1).")
t(X;6)(p22.3;21.1).")
Se informa a la paciente de dicho resultado y se decide continuar la gestación, realizando un control cada 2 semanas hasta la semana 36, pasando a controles semanales a partir de entonces. La biometría se lleva a cabo en cada control, con un PFE por debajo del percentil 3 y doppler normal en todos los controles, así como el perfil biofísico.
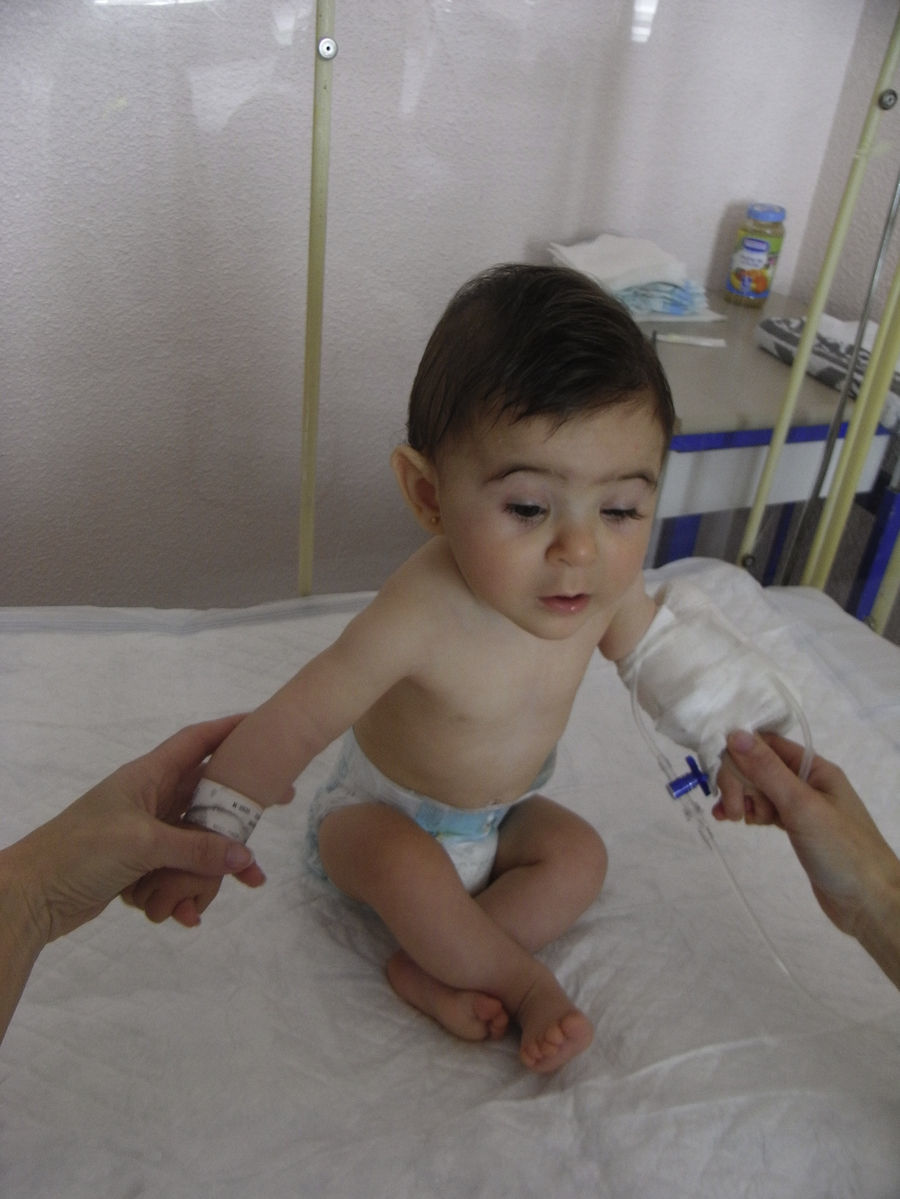
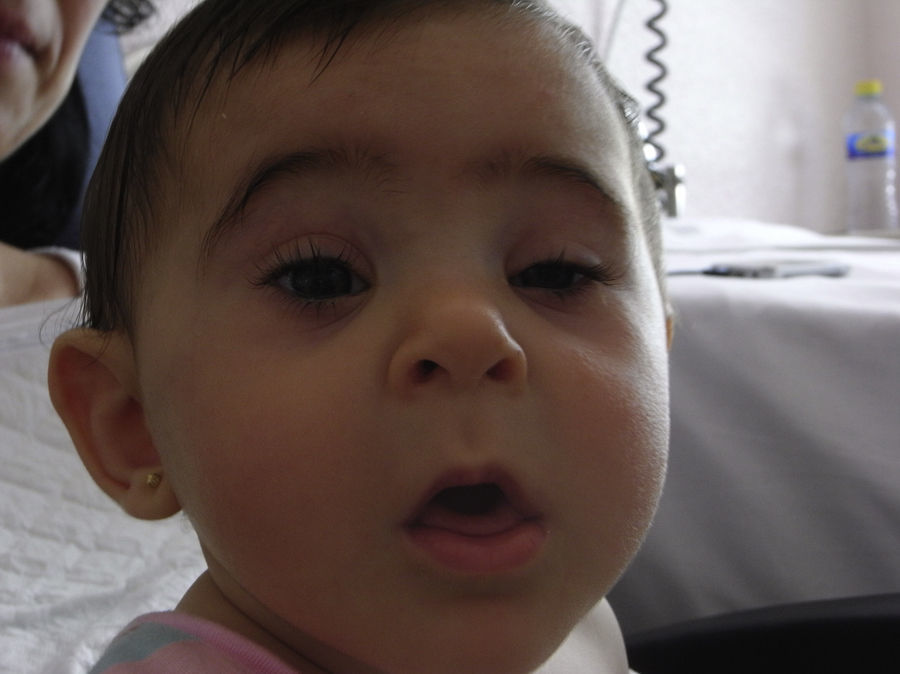
Se realiza una cesárea en la semana 39 por presentación podálica y riesgo de pérdida de bienestar fetal, naciendo una niña de 2.010 gramos (por debajo del percentil 3), apgar 9-10. La recién nacida presenta una facies dismórfica, con microftalmia, ptosis parpebral y quiste de cola de ceja izquierda. El resto de la exploración fue normal, excepto un peso por debajo del percentil 3. Las exploraciones complementarias fueron normales (ecografía cerebral, ecocardiografía, determinación de inmunoglobulinas) y la ecografía del quiste de la ceja informó de tumoración compatible con quiste dermoide. En la actualidad presenta un retraso severo del crecimiento y psicomotor, sin presentar ningún episodio de origen infeccioso (figs. 3 y 4).


Las alteraciones cromosómicas son una de las principales causas a descartar cuando se detecta un retraso del crecimiento intrauterino (PFE menor de un percentil 10). Están presentes en un 15% de los casos, sobre todo si se observa alguna malformación, si comienza antes de la semana 28 y si es una restricción severa (por debajo del percentil 3). Otras pruebas que deben realizarse son: estudio morfológico fetal, despistaje de preeclampsia (analítica, proteinuria en orina de 24h, control de la presión arterial). doppler de la arteria umbilical, arteria cerebral media y arterias uterinas, estudio infeccioso: TORCH y lúes (aunque las infecciones están presentes en menos del 5% de los casos) e incluso una amniocentesis para PCR de citomegalovirus si hay sospecha de infección por este virus. Si se produjera una preeclampsia o un desprendimiento previo de placenta normalmente inserta habría que realizar al finalizar la gestación un estudio de trombofilias.
En nuestro caso, el diagnóstico de alteración cromosómica se hizo por análisis de líquido amniótico, tras doble cultivo celular en monocapa, sacrificio celular y tinción convencional y bandas CTG. Se analizaron 20 metafases. El cariotipo fue una trisomía parcial del cromosoma 6 y una monosomía parcial del cromosoma X: 46X der(X)t(X;6)(p22.3;21.1). Los casos descritos en la literatura de trisomía parcial 6p21.1 a 6pter mostraron: retraso del crecimiento pre o posnatal, dificultad para la alimentación, rasgos dismórficos caracterizados por una facies plana, hipotelorismo, microftalmia, puente nasal amplio y prominente, y algunas malformaciones mayores como microcefalia, craneosinostosis, hidrocefalia, riñones hipoplásicos, hidronefrosis y malformaciones cardiacas entre otras1–3. Estos pacientes son propensos a las infecciones; casi la mitad de los pacientes descritos murieron durante el periodo de observación o durante los primeros meses de vida4.
Los casos de deleción parcial Xp22.3 a Xpter encontrados en la bibliografía mostraron microftalmia, agenesia del cuerpo calloso y malformaciones cardiacas como defectos más frecuentes6,7. Debido a que en el desbalance cromosómico está implicado el cromosoma X, no se puede saber con certeza la severidad de la repercusión fenotípica debido al mecanismo de inactivación del cromosoma X8,9.
El síndrome de MIDAS se caracteriza por la tríada microftalmia, esclerocórnea y aplasia dérmica7. Este síndrome se asocia en la mayoría de los casos con una microdeleción de Xp22.3, tratándose de un raro síndrome de herencia dominante ligada al cromosoma X, letal en varones. Las lesiones dérmicas típicamente afectan a cabeza, cuello y parte superior del tronco. Otras anomalías que pueden aparecer incluyen estatura corta, retraso psicomotor, defectos cardiacos congénitos, hernia diafragmática, agenesia del cuerpo calloso, anencefalia e hidrocefalia10. El caso que presentamos se ha catalogado como síndrome de MIDAS, ya que presenta la microdeleción de Xp22.3, microftalmia, esclerocórnea y otras anomalías asociadas (retraso de crecimiento y psicomotor) aunque hasta el momento no ha presentado ninguna lesión dérmica. Existen otros casos similares al nuestro en el que no se aprecian lesiones dérmicas pero sí la alteración cromosómica, con manifestaciones oculares11–13. Se ha sugerido que el mecanismo de inactivación del cromosoma X desempeñaría un papel importante en el desarrollo de algunos de los síntomas característicos del síndrome de MIDAS14,15.
La alteración cromosómica es heredada de la madre, que es portadora sana de una translocación recíproca entre los cromosomas X y 6, con puntos de corte en la banda p22.3 del cromosoma X y en la banda p21.1 del cromosoma 6. El estudio realizado es un cariotipo constitucional en sangre periférica mediante cultivo de linfocitos estimulados con fitohematoglutinina durante 72h, con tinción convencional y bandas GTG. Se analizaron 20 metafases Dado que se recomienda estudio de cariotipo en los familiares de primer grado, se solicitó estudio genético a la otra hija de la pareja, sana, de 7 años de edad, con resultado de cariotipo 46 XX normal.
Este caso de nuevo pone de manifiesto la importancia de investigar las alteraciones cromosómicas cuando se detecta un retraso del crecimiento intrauterino, sobre todo si es severo y precoz, para poder dar un consejo genético a la familia y un pronóstico fetal. Si el cariotipo fuera normal, se debería realizar un estudio array para detectar otras alteraciones genéticas que presentan retraso en el crecimiento intrauterino.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.