


En los últimos años, se ha producido un aumento preocupante en la prevalencia de la obesidad como consecuencia de los cambios en el estilo de vida, más sedentario y en los hábitos alimentarios que conducen a un aumento de la ingesta y por consiguiente a un desequilibrio energético. Las personas obesas tienen un mayor riesgo de desarrollar hipertensión, enfermedad coronaria, resistencia a la insulina, diabetes tipo 2 y síndrome metabólico. Además, el envejecimiento progresivo de la población, que suele ir acompañado de una disminución del ejercicio, aumento de la adiposidad y pérdida de musculatura, también contribuye a un aumento en el desarrollo de estas complicaciones. El excedente calórico se acumula en forma de triglicéridos en el tejido adiposo. No obstante, el exceso de lípidos también puede dar lugar a la acumulación de forma ectópica, en los tejidos no adiposos, contribuyendo así al daño de estos órganos mediante un proceso denominado lipotoxicidad. En este artículo se revisan los mecanismos de lipotoxicidad renal y como esta se ve afectada por la dislipidemia, el acúmulo de lípidos como consecuencia de la captación mediada por receptor, el transporte de lípidos facilitado por la albúmina y la síntesis de lípidos a nivel renal. El resultado es una alteración de metabolismo renal cuyos mecanismos incluyen la generación de especies reactivas del oxígeno, la alteración (y/o la parada) de rutas de señalización celular y la liberación de factores proinflamatorios y profibróticos que conducen a la lesión renal.
In recent years, there has been a worrying increase in the prevalence of obesity as a result of changes in lifestyle, such as greater sedentariness and modifications in dietary habits. These changes have led to an increase in food intake and energy imbalance. Obese patients are at greater risk of developing hypertension, heart disease, insulin resistance, type 2 diabetes and metabolic syndrome. Furthermore, progressive population aging, together with decreased exercise, increased adiposity and loss of muscle mass, also contributes to the development of these associated complications. The caloric excess accumulates in the form of triglycerides in adipose tissue. However, this excess can also accumulate in ectopic organs other than adipose tissue, which contributes to the damage of these organs through a process called lipotoxicity.
The present article reviews renal lipotoxicity and how it is affected by dyslipidemia, lipid accumulation resulting from receptor-mediated uptake, lipid transport facilitated by albumin, and lipid synthesis in the kidney. The result of this lipid accumulation is an alteration of renal metabolism whose mechanisms include the generation of reactive oxygen species, alteration and/or inactivation of cell signalling pathways, and the release of proinflammatory and profibrotic factors, leading to renal disease.
En las últimas dos décadas, el mundo occidental ha sufrido un aumento preocupante en la prevalencia de la obesidad, como consecuencia de un estilo de vida sedentario y un cambio en los hábitos alimentarios. Las personas obesas tienen un mayor riesgo de desarrollar hipertensión, enfermedad coronaria, resistencia a la insulina, diabetes tipo 2 y síndrome metabólico. Además, estudios recientes asocian la enfermedad crónica renal con la obesidad. Esta problemática sumada a la lesión renal asociada al envejecimiento progresivo de la población y al aumento en la esperanza de vida, suponen un incremento del gasto del sistema sanitario de grandes dimensiones.
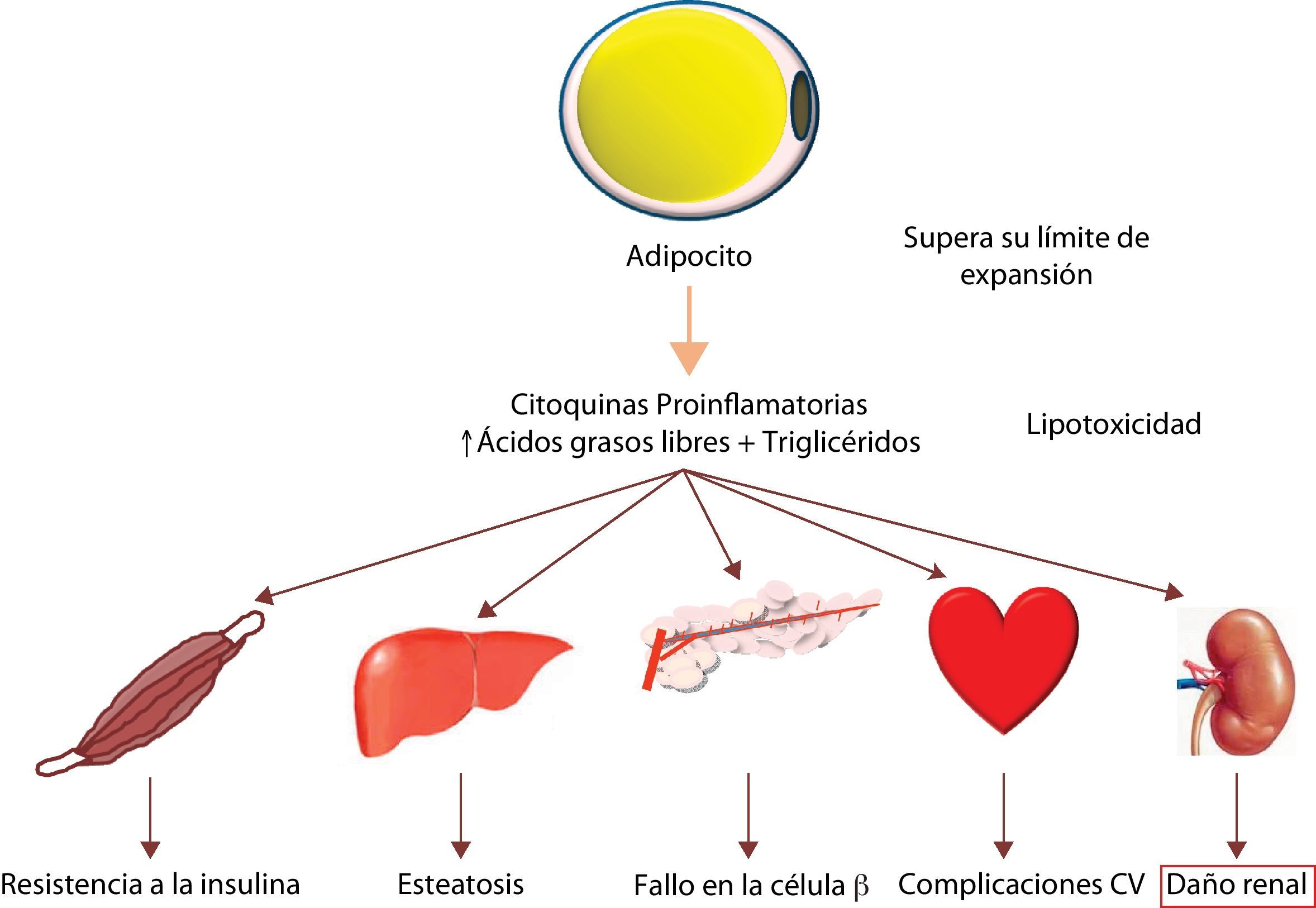
El exceso de lípidos que se acumulan en los tejidos no adiposos contribuye al daño de estos órganos mediante un proceso denominado lipotoxicidad (fig. 1). La lipotoxicidad se produce por un exceso en el contenido intracelular de ácidos grasos no esterificados y por la acumulación de sus derivados tóxicos, como los diacilgliceroles y las ceramidas. La disfunción y el daño celular provocado por la lipotoxicidad se producen a través de varios mecanismos incluyendo la generación de especies reactivas del oxígeno, la alteración (y/o la parada) de rutas de señalización celular, la liberación de factores proinflamatorios y profibróticos. Estos conducen al daño en diversos orgánulos celulares y a la inducción de apoptosis por lípidos, también llamada lipoapoptosis1. Este proceso se ha descrito en múltiples tejidos incluyendo las células musculares y miocitos, los hepatocitos y las células beta-pancreáticas2–5.

En 1858, Virchow sugería, por primera vez, la existencia de una asociación entre la presencia de lípidos y el desarrollo de enfermedad renal. Sus primeros trabajos describían una serie de etapas sucesivas de metamorfosis de la grasa, así como la presencia de acúmulos lipídicos en el epitelio renal en la enfermedad de Bright6. Más tarde en 1936, Kimmelstiel y Wilson7 describieron los signos patológicos de la esclerosis nodular, y además demostraron la presencia de depósitos lipídicos en el riñón de pacientes diabéticos. Ya en aquellos años sus estudios sugerían el importante papel de los lípidos en la patogénesis de la enfermedad renal. En los últimos años, existen evidencias que sugieren que la acumulación renal de lípidos y la lipotoxicidad pueden conducir a la disfunción del riñón. Apoyando esta hipótesis, se ha visto que el tratamiento con inhibidores de la β-hidroximetil glutaril Co A (HMG-CoA) reductasa, la principal enzima reguladora de la síntesis de colesterol, mejora la proteinuria y preserva la función renal independiente de otras variables, lo que sugiere un papel de los lípidos, per se, en la promoción de la lesión renal8–10.
El riñón y el metabolismo lipídicoUn riñón humano tiene alrededor de un 3% de materia grasa, existiendo una gran variabilidad entre individuos. La mitad aproximadamente de esa grasa son fosfolípidos que forman parte de las membranas, un 15% son triglicéridos y en torno a un 10% son ácidos grasos libres no esterificados11.
El riñón, en condiciones fisiológicas, es capaz de captar de la sangre que circula por las arterias una gran variedad de sustratos y utilizarlos como combustible: ácidos grasos libres, lactato, glutamina, 3-hidroxibutirato, citrato, piruvato, α-cetoglutarato, glicerol, prolina, y otros aminoácidos que se encuentran en menor cantidad12. El túbulo proximal es responsable de reabsorber el 70% de estos sustratos. El destino metabólico de esta mezcla de sustratos depende del medio extracelular, las influencias hormonales y las condiciones metabólicas (ácido-base)13.
Una vez reabsorbidos, los ácidos grasos libres son β-oxidados en el interior de la mitocondria de las células del túbulo proximal y constituyen la mayor fuente de producción de ATP, ya que su capacidad glicolítica es bastante baja13. Cuando existe un aumento de la concentración intracelular de ácidos grasos, se produce una competición con el resto de los sustratos oxidables por la mitocondria. Este hecho desencadena un descenso en la utilización de glutamina, con la consiguiente reducción en la producción de amonio. Esto podría explicar por qué los obesos y las personas con síndrome metabólico tienen mayor riesgo de desarrollar una nefrolitiasis ácido úrico idiopática, caracterizada por un descenso en el pH de la orina1.
Los ácidos grasos libres no esterificados circulan por el plasma unidos a la albúmina y solamente una porción mínima (menor del 0,01%) se encontraría libre. El riñón tiene gran capacidad de captar esta albúmina unida a lípidos y otras proteínas transportadoras de lípidos, proteínas reguladas por lípidos y hormonas (como por ejemplo la apolipoproteína AI y leptina). Esta absorción se produce, fundamentalmente, a través de receptores que se encuentran en la superficie apical de los túbulos proximales: megalina y cubilina/receptor de la proteína amnionless. A pesar de que la barrera de filtración glomerular impide el acceso de las grandes partículas lipoproteicas a los túbulos proximales, los receptores pueden estar expuestos a los lípidos que se encuentran unidos a las proteínas filtradas. En consecuencia, el metabolismo lipídico sistémico puede verse influenciado por la filtración y la captación de lípidos mediada por receptor que ocurre en el riñón14.
Por otra parte, se ha visto que el riñón no solo es capaz de captar lípidos, sino que también puede sintetizar apolipoproteína B15 y apolipoproteína E16, y excretarlas al torrente sanguíneo en forma de lipoproteínas, disminuyendo de esta manera el exceso de triglicéridos que llegan al túbulo proximal. Existen autores que hipotetizan que este proceso sería una forma de recuperar componentes lipofílicos (vitaminas) a la circulación sanguínea, que hubieran sido filtrados en la orina primaria14.
Los lípidos en la fisiopatología renalFactores implicados en la lipotoxicidad renalLos estudios realizados hasta la fecha indican que existen varios factores implicados en la lipotoxicidad renal y la consecuente disfunción del riñón.
En primer lugar, el riñón se ve afectado por la dislipidemia, es decir por una alteración en los niveles de lípidos y lipoproteínas en sangre. En 1982, Moorhead establecía por primera vez la idea de que la dislipidemia podía contribuir a la enfermedad renal17. Hoy en día, se sabe que la dislipidemia puede actuar directamente sobre el riñón. Esta alteración causa un efecto deletéreo debido a la acumulación de ácidos grasos libres no esterificados y, de forma indirecta debido a la inflamación sistémica, el aumento del estrés oxidativo (EO) y la producción y activación de citoquinas y hormonas asociadas a la fisiopatología renal. Estudios en la población en general, han asociado la presencia de hiperlipidemia con el desarrollo de enfermedad crónica renal en la que está implicado tanto el EO como el estrés del retículo endoplásmico (RE). El EO se manifiesta por un aumento de especies reactivas de oxígeno (ROS), consecuencia de un desequilibrio entre los sistemas que las producen como la actividad mitocondrial o actividades como NADPH oxidasa (NOX) y los sistemas que las eliminan como superóxido dismutasa o caltalasa18. Las ROS, además de alterar una gran variedad de estructuras celulares debido a su reactividad química, también inducen respuestas inflamatorias19. El estrés del RE se produce por una disrupción del plegamiento proteico y la subsiguiente activación de rutas específicas de comunicación entre el RE, el citoplasma y el núcleo, las cuales están asociadas a la patogénesis de muchas enfermedades, incluyendo la diabetes20,21. En estas enfermedades, este estrés de RE desencadena lo que se conoce como unfolded protein response (UPR) que incluye diversas respuestas a nivel transcripcional como el aumento de la síntesis de chaperonas, la inhibición de la síntesis de proteínas y la activación de la degradación de proteínas vía proteasoma, todas ellas encaminadas a reestablecer el equilibrio22.
En segundo lugar, la disfunción renal es consecuencia de la acumulación de lípidos que ocurre tanto a nivel glomerular como a nivel tubular, fundamentalmente en el segmento proximal. Esta acumulación de lípidos en el riñón está asociada a cambios en la expresión de genes tan importantes en la regulación del metabolismo lipídico como el sterol regulatory element binding protein (SREBP) y también a la activación de la ruta de transforming growth factor-β (TGF-β) por la inducción de ROS23,24. Estos cambios en el metabolismo lipídico intrarrenal favorecen el daño renal y ponen de manifiesto que el riñón no actúa como un órgano pasivo que se ve afectado únicamente por los cambios que ocurren a nivel sistémico.
En tercer lugar, cabe destacar que la albúmina juega un papel fundamental en el daño renal mediado por lípidos, ya que puede actuar como un transportador pasivo, como un «caballo de Troya» de ácidos grasos libres no esterificados en el túbulo proximal25. Como hemos comentado anteriormente, los ácidos grasos no esterificados se transportan, mayoritariamente, en el plasma a través de la albúmina. Se ha observado que, cuando hay un exceso de ácidos grasos, estos se depositan en el riñón en forma de gotas lipídicas que pueden ser detectadas mediante la tinción Oil Red, tanto en el hombre como en animales. Se ha propuesto que el exceso de ácidos grasos no esterificados en el túbulo proximal puede ser consecuencia de un aumento en la filtración de albúmina que transporta lípidos y a la vez a un incremento en la tasa de lípidos transportados por unidad de albúmina26,27. Este proceso conduce a un aumento en la concentración intracelular de ácidos grasos en las células tubulares que excede la capacidad β-oxidativa de la mitocondria y tiene como resultado una acumulación de triglicéridos y la generación de metabolitos lipídicos con efecto potencialmente tóxico como las ceramidas, que además pueden interferir con mecanismos de señalización celular como el de la insulina, causando resistencia a dicha hormona.
Por último, existen una serie de estudios recientes acerca del efecto lipotóxico sobre los podocitos y la barrera de filtración glomerular. Diversos estudios muestran como segmentos ricos en colesterol (rafts lipídicos) se insertan en el diafragma de rendija de los podocitos entre la podocina y la nefrina que ayudan a mantener la unión de podocina-nefrina y nephrin-transient receptor potencial channel 6 (TRPC-6). Además, la podocina sufre una modificación postraduccional, una palmitoilización, que regula su interacción de forma precisa con los rafts lipídicos. La desregulación de la oxidación de los ácidos grasos puede afectar a la palmitoilización de la podocina y por tanto, a la correcta inserción en la membrana. Una sobrecarga de ácidos grasos puede alterar la composición lipídica de los rafts e interferir en la señalización de la red podocina-nefrina-TRPC-6-actina del citoesqueleto produciendo una cascada de efectos patológicos28.
Los lípidos en la nefropatía diabéticaLa nefropatía diabética (ND) está aumentando de forma preocupante a nivel mundial, como causa de morbimortalidad. Alrededor de un tercio de los pacientes diabéticos desarrollan nefropatía, por lo que resulta especialmente importante el diagnóstico precoz para evitar la pérdida de la función renal. Además, la patología de la ND asociada a la diabetes tipo 2 está a menudo complicada con el síndrome metabólico asociado a la diabetes tipo 2, en el que la hiperlipidemia, la obesidad y la hipertensión pueden conjuntamente afectar a la progresión de la enfermedad renal.
La ND se caracteriza por un gran número de cambios estructurales a nivel histológico y cambios a nivel celular que incluyen la secreción de múltiples factores y citoquinas que establecen un ciclo vicioso al promover infiltración de macrófagos e inflamación que hace que la enfermedad empeore y avance más rápidamente.
A nivel glomerular, se produce un incremento del tamaño del glomérulo por efecto de la expansión mesangial y de la hiperotrofia compensatoria. Los cambios en la expresión de sustancias vasoactivas provocan dilatación arteriolar aferente y en consecuencia, una hiperperfusión glomerular. En estas circunstancias fisiológicas, el mantenimiento de la barrera de filtración glomerular depende de la secreción, la degradación y la capacidad contráctil de los podocitos. La hiperglucemia, a través de la angiotensina II (AngII), induce el aumento de la expresión de PTHrP en los podocitos, la cual induce la expresión de TGF-β y del inhibidor 1B de quinasa dependiente de ciclina, p27(Kip1)29. La hiperglucemia en podocitos también induce la síntesis del vascular endothelial growth factor (VEGF)30. El VEGF estimula las células endoteliales, que sintetizan platelet-derived growth factor (PDGF). Este factor a su vez activa las células mesangiales, produciendo un incremento en la síntesis de proteínas de la matriz (colágeno tipo IV y fibronectina). El TGF-β, también, disminuye la expresión de la nefrina en podocitos, una proteína clave que junto a proteínas transmembrana forma parte de las rendijas de filtración que existen entre los pedicelos de los podocitos31. Además, estudios muy recientes en podocitos, muestran que no solo la hiperglucemia sino también la señalización de insulina en estas células es crucial para el remodelado del citoesqueleto de actina y el mantenimiento de la integridad de la barrera de filtración glomerular32. En las células mesangiales, la hiperglucemia y los productos avanzados de glicosilación (AGE) inducen la secreción de monocyte chemoattractant protein-1 (MCP1), lo que produce la infiltración de macrófagos en el glomérulo y la consiguiente inflamación. Las células endoteliales también se ven afectadas por la hiperglucemia, ya que se ha visto que durante la diabetes reducen la producción de eicosanoides: los derivados del ácido araquidónico que actúan como sustancias glomeruloprotectoras. Este descenso en eicosanoides favorece el aumento de la permeabilidad del endotelio33.
A nivel tubular, la progresiva glomeruloesclerosis induce una isquemia crónica. Por otra parte, el aumento de la permeabilidad de la barrera de filtración glomerular produce un incremento de sustancias filtradas y de citoquinas en el túbulo contorneado proximal y en los capilares intertubulares e intersticio que lo rodea. Además, las células tubulares son capaces de responder a la hiperglucemia, el EO, la albuminuria, la AngII y los lípidos, con la consiguiente secreción de citoquinas. El resultado es que a nivel tubular se produce hipertrofia como mecanismo compensatorio del aumento del volumen de filtrado glomerular. Por último, en el intersticio se produce una inflamación y fibrosis, como consecuencia de las alteraciones del flujo sanguíneo y la secreción de múltiples factores y citoquinas por las células colindantes34.
En pacientes diabéticos el exceso de lipoproteínas y lípidos acelera la progresión de la ND, ya que aumenta el daño glomerular y la fibrosis tubulointersticial. Además, estos pacientes presentan en plasma unos niveles elevados de VLDL, IDL y LDL, y concentraciones bajas de colesterol HDL. Parece ser que por un lado, los lípidos provocan daño renal por la estimulación de TGF-β, y la consecuente inducción de especies reactivas de oxígeno y daño en los glomérulos y glicocálix glomerular. Por otro lado, las lipoproteínas ricas en triglicéridos pueden activar monocitos y degradar el glicocálix. Este hecho aumenta la permeabilidad de la barrera de filtración glomerular y contribuye a la progresión de la nefropatía diabética34.
La diabetes está asociada a alteraciones de distintas clases de apolipoproteínas (apo), cuyas acciones específicas pueden afectar al desarrollo de la nefropatía en pacientes diabéticos. Así, se ha descrito que pacientes diabéticos presentan niveles elevados plasmáticos de apoB48, una proteína estructural de los quilomicrones35, apoC-I, apoC-III/apoE36. También, se ha descrito que individuos obesos con enfermedad renal presentan elevados niveles plasmáticos de insulina y apoC-III/apoE, por lo que se sugiere que apoC-I, apoC-III/apoE están asociados a la enfermedad renal37,38. Las lipoproteínas en pacientes diabéticos son glicosiladas de forma no enzimática y esas LDL y VLDL glicosiladas son más susceptibles de ser oxidadas que las formas no glicosiladas. Por otra parte, se han descrito niveles altos de LDL oxidados (Ox-LDL) en pacientes diabéticos con macroalbuminuria, por lo que se asocia este aumento con la progresión de la nefropatía diabética39. Las Ox-LDL inducen un descenso en la expresión de nefrina en podocitos en cultivo. La nefrina una vez fosforilada se asocia con phosphatidylinositol 3-kinase (PI3K) y estimula la ruta de señalización dependiente de protein kinase (Akt) B. Esta ruta juega un papel crucial en el reordenamiento y la activación dependiente de actina-nefrina del citoesqueleto implicada en el control del tráfico de proteínas y en la supervivencia de los podocitos.
Cuando se estudia el efecto de los lípidos en modelos animales de diabetes (ratas STZ, ratones db/db, ratas ZDF) nos encontramos con que existe una acumulación de lípidos tanto a nivel tubular como glomerular, acompañado de una sobreexpresión de SREBP40,41. Proctor et al. (2006)42 proponen que durante la diabetes, el metabolismo renal experimenta un cambio global, que se traduce en 1) un aumento en la síntesis de ácidos grasos mediada por un incremento en SREBP-1, acetil-CoA carboxylase (ACC), fatty acid syntase (FAS), stearoyl-CoA desaturase 1 (SCD-1), carbohydrate responsive element-binding protein (ChREBP) y L-type pyruvate kinase (L-PK); 2) un descenso en la oxidación de los ácidos grasos mediada por una bajada en la expresión de peroxisome proliferator-activated receptors α y δ (PPAR-α, PPAR-δ), y acyl-CoA oxidase (ACO); 3) un aumento en la síntesis de colesterol mediada por un incremento en SREBP-2 y HMG-CoA reductasa; y 4) un descenso del flujo de colesterol mediado por una disminución en liver X receptor α y β (LXR-α, LXR-β), y ATP-binding cassette transporter-1 (ABCA-1). Además, en estos animales aparecen aumentados los niveles de factores de crecimiento profibróticos (TGF-β, plasminogen activator inhibitor-1 [PAI-1] y VEGF), los niveles de citoquinas proinflamatorias (interleuquina 6 [IL-6] y TNF-α); el EO y los receptores de productos finales de glicosilación avanzada (RAGE).
Efecto de la obesidad en el desarrollo de enfermedad renalEn 1974, se estableció por primera vez una asociación entre la obesidad y el síndrome nefrótico43,44. En los últimos años, se ha visto que la glomerulopatía relacionada con la obesidad y las enfermedades crónicas del riñón son complicaciones propias de la obesidad. Concretamente, los efectos renales de la obesidad presentes en humanos y en animales de experimentación incluyen adaptaciones estructurales y funcionales, como el aumento de la tasa de filtración glomerular, el aumento del flujo sanguíneo renal, la hipertrofia renal, glomeruloesclerosis segmental y glomerulomegalia45.
En modelos de animales obesos46,47 se ha descrito que existe una acumulación lipídica a nivel glomerular, intersticial y en las células del túbulo proximal, acompañada de un aumento en la expresión de los SREBP renales y otros genes lipogénicos. El tratamiento con agonistas de PPARγ atenúa esta acumulación y disminuye el daño mediado por lípidos a nivel renal. Este mismo efecto de atenuación ocurre en los modelos de ratones heterocigotos para PPARγ, en los que se induce un daño renal por una dieta con alto contenido en grasas46. Por otra parte, en el ratón knock-out para SREBP-1c alimentado con una dieta rica en grasa se induce una elevación del contenido renal de triglicéridos y un incremento en la expresión de PAI-1, VEGF y de las proteínas de la matriz extracelular, en contraste con el ratón control. Sugiriéndose, que la obesidad inducida por una dieta rica en grasa causa aumento de la acumulación renal de lípidos y glomeruloesclerosis, vía una ruta dependiente de SREBP-1c48.
Además, desde que se describiera la leptina como un factor saciante derivado de los adipocitos, el tejido adiposo se ha empezado a considerar también como un órgano endocrino.
Así, se ha visto que la leptina induce la proliferación de células endoteliales en el glomérulo y un aumento de la expresión de TGF-β y de colágeno tipo IV en animales, lo que conduce a una glomeruloesclerosis y a la aparición de proteinuria49. Este mecanismo podría ser relevante en individuos obesos con niveles altos de leptina en sangre. Por otro lado, existen estudios que muestran la existencia de una correlación negativa en pacientes obesos entre los niveles de adiponectina y la proteinuria50. En este sentido se ha observado que el ratón knock-out para adiponectina presenta unos niveles basales de albuminuria dos veces superiores al normal y los pedicelos de los podocitos se encuentran afectados50.
Envejecimiento, lípidos y alteración renalEl envejecimiento es un complejo proceso multifactorial asociado a numerosas alteraciones anatómicas y funcionales. Concretamente, el riñón sufre una serie de procesos involutivos entre los que se incluyen la progresiva pérdida de masa renal, asociada a una pérdida de nefronas funcionales, a la aparición de esclerosis glomerular y al aumento de la fibrosis intersticial medular. Así también, con la edad se observa una disminución gradual del flujo sanguíneo renal (FSR) a un ritmo aproximado de un 10% por década y una disminución de la tasa de filtración glomerular (TSG), probablemente provocada por una mayor permeabilidad de la membrana glomerular y una menor superficie disponible de filtración.
En los modelos animales de envejecimiento48,51 se ha encontrado que los ratones C57BL/6 presentan con la edad un aumento progresivo de glomerulosclerosis determinado como un aumento en la tinción periodic acid-Schiff (PAS) y una acumulación de proteínas de la matriz extracelular, como el colágeno tipo IV y la fibronectina. Así también, estos ratones muestran un mayor grosor de la membrana basal glomerular, un ensanchamiento de los podocitos y en consecuencia, un aumento en la proteinuria. Pero además, estos cambios relacionados con la edad, están asociados a un incremento en la acumulación de lípidos en el riñón, concretamente al contenido renal de triglicéridos y colesterol. También se ha encontrado un aumento significativo en el riñón de los factores de transcripción nuclear SREBP-1 y SREBP-2 y de enzimas implicadas en la síntesis de ácidos grasos y colesterol. Asimismo, se ha mostrado que el desarrollo de enfermedad renal como consecuencia de la edad puede ser corregido, en parte, por la restricción calórica51. Esta restricción supone una modulación tanto de la expresión renal de SREBP, como de la acumulación de lípidos en el riñón.
En 1995, los estudios de Siest et al. mostraron que los polimorfismos de la apoE son un factor de riesgo para la patogénesis de enfermedades relacionadas con la edad52. La apoE es una proteína sintetizada principalmente por el hígado, aunque también la producen otros órganos o tejidos como el riñón, el cerebro, el bazo y los macrófagos y es crítica para la formación y estabilidad de los quilomicrones, las lipoproteínas de muy baja densidad (VLDL) y las lipoproteínas de alta densidad (HDL) Recientemente, los estudios en el ratón knock-out apoE muestran que este ratón presenta alteraciones morfológicas y bioquímicas dependientes de la edad, tales como la fibrosis, citoquinas proinflamatorias (IL-6 e iNOS), acumulación de lipofuscina y disminución de enzimas antioxidantes en diferentes órganos (riñón, hígado y corazón), que no se encuentran en animales control53,54.
Relación entre hipertensión y depósitos de lípidos en el tejido renalLa hipertensión está estrechamente vinculada a la obesidad y es, probablemente, una de las principales causas de la disfunción renal de los pacientes obesos, pero no es probable que sea la única causa hemodinámica55. El aumento del tono vascular y la retención de agua y sal por parte del riñón son los principales iniciadores de la hipertensión en la obesidad. Los mecanismos subyacentes incluyen hiperleptinemia, aumento de los ácidos grasos libres, hiperinsulinemia y resistencia a la insulina, que causa la estimulación del sistema nervioso simpático, aumento del tono vascular, disfunción endotelial, y retención renal de sodio. Además, el aumento de la actividad del sistema renina-angiotensina (RAS), como resultado tanto de la activación simpática como de, posiblemente, el aumento de la generación del tejido adiposo, se traduce en un aumento de sodio renal y una retención de agua56.
El estudio en ratas con hipertensión inducida con infusión de AngII mediante minibomba subcutánea57, muestra que se producen depósitos de lípidos en las células del túbulo renal acompañados de un aumento en la producción de superóxido, en la expresión de TGF-β, SREBP-1 y de la ácido graso sintasa (FAS), respecto a las ratas control. El tratamiento de estas ratas infundidas con un agente quelante del hierro, deferoxamina, atenúa el incremento de la expresión renal de SREBP-1, de la FAS y normaliza el contenido lipídico del tejido cortical renal. También, se ha visto que la infusión de AngII induce el aumento de la expresión del ARNm de PDGF-β y PDGFR-β en el riñón, y este efecto puede ser suprimido tratando a los animales con losartán, un antagonista del receptor AT1 de AngII58. En el riñón de estas ratas infundidas con AngII, las células que tienen aumentada la expresión de PDGF-β son precisamente aquellas que acumulan lípidos y también las que son proliferating cell nuclear antigen (PCNA) positivas. Lo que sugiere que la deposición lipídica, el PDGF y la proliferación celular renal pueden estar relacionadas en la hipertensión inducida por AngII en ratas58. Por otra parte, las ratas espontáneamente hipertensas (SHR) alimentadas con dieta rica en grasa muestran una acumulación de triglicéridos y ácidos grasos libres asociados a cambios estructurales renales (glomerulosclerosis, inflamación y apoptosis). Estos animales son incapaces de incrementar la expresión proteica de PPARα en el riñón. Sin embargo, el tratamiento con fenofibrato, un ligando de PPARα, reduce la acumulación lipídica, la inflamación y en general, el daño renal en este modelo animal de hipertensión y obesidad59.
Nuevas terapias en el tratamiento del daño renal asociado a alteraciones lipídicasEn 1986, se publicó un estudio en el que se mostraba que la dieta rica en aceite de pescado reducía la progresión del fracaso renal ya establecido60. Los últimos estudios en ratas nefrectomizadas muestran que la suplementación de la dieta con el ácido graso omega -3 (O-3) atenúa la lesión renal tubulointersticial y la fibrosis en el riñón remanente. En estas ratas nefrectomizadas suplementadas con O-3 se observa una reducción significativa de la expresión de TGF-β, mothers against DPP homolog 2 (Smad2), connective tissue growth factor (CTGF), alpha Smooth muscle actin (α-SMA), PAI-1 y extracellular-signal-regulated kinases (ERK) 1/2 fosforilado y la restauración parcial de Smad761.
Otra terapia que se ha probado es la administración de fibratos, derivados del ácido fibrico y agonistas de PPARα, que producen un descenso del contenido en triglicéridos y colesterol en plasma y que se usan frecuentemente en el tratamiento de la hiperlipidemia. Sin embargo, el uso de fibratos en pacientes con insuficiencia renal media o moderada provoca un deterioro de la función renal como consecuencia del incremento de creatinina sérica. El mecanismo implicado en este aparente deterioro de función renal secundario al tratamiento con fibratos no ha sido aún esclarecido62,63.
El tratamiento con estatinas está recomendado para la prevención primaria y secundaria de la enfermedad vascular en un gran número de pacientes y ensayos64,65. Los últimos estudios han demostrado que el tratamiento con estatinas produce un amplio beneficio en individuos con enfermedad crónica renal no dependiente de diálisis66. El macroestudio Study of Heart and Renal Protection (SHARP) sugiere que este tratamiento es beneficioso incluso para los pacientes en diálisis67,68.
Además de los agonistas de PPARα, se ha ensayado el tratamiento con tiazolidinedionas (TZD), agonistas de PPARγ. El efecto beneficioso que producen en la enfermedad renal se debe a que estos fármacos mejoran la tolerancia a la glucosa y en consecuencia, indirectamente mejoran la progresión de la enfermedad renal. Junto a los anteriores estudios, en los últimos años se ha visto que los antiproteinúricos, antiinflamatorios y antifibróticos tienen efectos directos sobre el riñón los cuales son independientes de la mejora de la tolerancia a la glucosa69.
No obstante, a la espera de nuevos datos, creemos que sería necesario el desarrollo de nuevos estudios farmacológicos que disminuyan la lipotoxicidad y la inflamación para prevenir y/o tratar la enfermedad renal asociada al síndrome metabólico y al envejecimiento.
Conflicto de interesesLos autores declaran que no tienen ningún conflicto de intereses.