







El carcinoma familiar de tiroides no medular (FNMTC, familial non-medulary thyroid carcinoma) se define por la presencia de 2o más miembros familiares de primer grado con carcinoma diferenciado de tiroides (CDT). El objetivo de este estudio es analizar las características clínicas, anatomopatológicas y pronósticas del FNMTC respecto al carcinoma esporádico (CE).
Materiales y métodosEstudio retrospectivo de los casos de CDT registrados en nuestro centro hospitalario durante el periodo 1990-2018.
ResultadosSe analizó a un total de 927 pacientes, de los cuales 61 son FNMTC, con un seguimiento medio de 9,7± 6,5 años. La prevalencia del FNMTC es del 6,6%, presentando un estadio TNM inicial más bajo (p=0,003) debido a la mayor proporción de tumores inferiores a 2 cm (p=0,003), además de ser más multifocales (p=0,034) y del subtipo histológico papilar (p=0,022), respecto al CE. No se detectaron diferencias significativas en la edad al diagnóstico (p=0,347), género (p=0,406), ni en otros marcadores de agresividad (bilateralidad, extensión extratiroidea, afectación ganglionar y metástasis). La tasa de persistencia/recurrencia (p=0,656), supervivencia libre de enfermedad (p=0,929) y mortalidad ocasionada por el propio tumor (p=0,666) fueron comparables. En las familias con ≥ 3 familiares afectados, los tumores son más pequeños (p=0,005), más multifocales (p=0,040) y bilaterales (p=0,002), además de haber una mayor proporción de hombres (p=0,020). Los pacientes de la segunda generación presentaron al diagnóstico el FNMTC a una edad más precoz respecto a los de la primera (p=0,001).
ConclusiónEl FNMTC presenta en nuestro estudio, en comparación con el CE, una estadificación TNM más baja, mayor multifocalidad y representación del subtipo papilar, con similares datos de agresividad y sin diferencias en el pronóstico.
Familial non-medullary thyroid carcinoma (FNMTC) is defined by the presence of 2or more first-degree family members with differentiated thyroid carcinoma (DTC). The aim of this study is to compare clinicopathological features and prognosis of FNMTC and sporadic carcinoma (SC).
Materials and methodsRetrospective study of DTC included in the hospital database during the period 1990-2018.
ResultsA total of 927 patients were analyzed, 61 of them were FNMTC, with a mean follow-up of 9.7±6.5 years. The prevalence of FNMTC was 6.6%, with a lower TNM staging presentation (P=.003) consequence of a higher proportion of tumors smaller than 2 centimeters (P=.003), combined with a greater multifocality (P=.034) and papillary histologic subtype (P=.022) compared to SC. No significant differences in age at diagnosis (P=.347), gender (P=.406), neither in other aggressiveness markers (bilaterality, extrathyroidal extension, lymph node involvement and metástasis) were detected. Rate of persistence/recurrence (P=.656), disease-free survival (P=.929) and mortality caused by the tumor itself (P=.666) were comparable. Families with ≥3 affected relatives, had smaller tumors (P=.005), more multifocality (P=.040) and bilaterality (P=.002), as well as a higher proportion of males (P=.020). Second generation patients present earlier FNMTC compared to those of the first generation (P=.001).
ConclusionIn our study FNMTC presents a lower TNM staging, higher multifocality and papillary variant, with similar aggressiveness and prognosis compared to SC.
El carcinoma de tiroides (CT) supone el 1% de todos los tumores, siendo la neoplasia endocrina más frecuente con una incidencia creciente en las últimas décadas en numerosos países a nivel mundial1,2. Sin embargo, la mortalidad se ha mantenido estable en un 0,6% por año, ya que la mayoría son tumores subclínicos (carcinomas papilares inferiores a los 2cm)3,4. Los distintos subtipos de tumores tiroideos se clasifican atendiendo a su origen histológico. Más del 90% de los carcinomas derivan de las células foliculares y se denominan carcinoma diferenciado de tiroides (CDT), que engloba los subtipos papilar, folicular y carcinoma de células de Hürthle5. Una minoría proviene de las células parafoliculares, originando el carcinoma medular de tiroides.
Existe una agregación familiar en los CDT no medulares de aproximadamente el 5-10%5. Por ello, se ha definido como carcinoma familiar de tiroides no medular (FNMTC, familial non-medulary thyroid carcinoma) aquellos carcinomas bien diferenciados con origen en las células foliculares que se presentan en 2o más familiares de primer grado, en ausencia de otros factores de predisposición hereditarios o ambientales conocidos6. Pueden coexistir distintas variantes histológicas en la misma familia. Algunos autores son más conservadores y para no sobrestimar la prevalencia debida al azar, sugieren que sean al menos 3los familiares afectados7. En esta línea, un análisis matemático calculaba que la probabilidad estimada de que el CDT fuese esporádico era del 62-69% si se contaba con 2familiares diagnosticados, disminuyendo hasta el 6% si eran 3o más los familiares afectados8. En este caso, el riesgo de desarrollar CDT en familiares de primer grado de pacientes diagnosticados de CT puede multiplicarse en más de 3veces9,10. El cribado ecográfico ha demostrado detectar CDT hasta en un 22,7% de los casos en familias con 3o más miembros afectados y tan solo 4,6% en familias de 2, por lo que podría aportar beneficio en el primer grupo. Para realizar este cribado es importante tener en cuenta el fenómeno de anticipación en la segunda generación, que desarrolla la enfermedad a edad más precoz y de una forma más avanzada al diagnóstico11.
Las bases genéticas de estos síndromes son complejas. El 5% de los FNMTC se presentan como parte del espectro de síndromes hereditarios (síndrome de Gardner, síndrome de Cowden, complejo de Carney, síndrome de DICER1, síndrome de Werner) y se denomina FNMTC sindrómico12. Sin embargo, el 95% de los casos restantes ocurren de forma aislada, denominándose FNMTC no sindrómico. Las causas genéticas de estos últimos están aún por dilucidar. La evidencia actual va a favor de mecanismos de herencia poligénicos, siendo una minoría los carcinomas debidos a mutaciones localizadas en genes de susceptibilidad13. Se ha demostrado que estos pacientes tienen un incremento en la inestabilidad de los telómeros en comparación con sujetos sanos y cáncer esporádico, fenómeno implicado en una mayor tendencia al desarrollo de cáncer14. Estudios recientes han identificado un panel de genes asociados a mayor vulnerabilidad para el desarrollo del FNMTC, que son los siguientes: FOXE1, TPCSC2, MYH9, SRGAP1, HABP2, BRACA1, CHEK2, ATM, RASAL1, SRRM2, XRCC1, TIRT-1/NKX2.1 y PTCSC315. Sin embargo, los resultados obtenidos hasta la fecha no permiten determinar todavía el impacto de estos genes en el desarrollo y el pronóstico del FNMTC, por lo que hoy en día no está indicado el estudio genético de las familias.
Se han llevado a cabo numerosos estudios comparando a los pacientes con FNMTC frente al carcinoma esporádico (CE), posicionándose a favor de una mayor agresividad en el grupo familiar16,17. Sin embargo, su forma de presentación y pronóstico son aspectos controvertidos, dada la heterogeneidad de los estudios y su diseño retrospectivo18. Nuestro estudio tiene como objetivo principal comparar las características clínicas y anatomopatológicas de agresividad entre el FNMTC no sindrómico y el CE. También se analizaron las diferencias existentes en el subgrupo de familias con 2y 3miembros afectados de CDT respecto a aquellos con CE, así como de los miembros de primera frente a los de segunda generación familiar. Por último, se pretende valorar las diferencias en el tratamiento inicial y determinar el pronóstico del FNMTC confrontado con el CE.
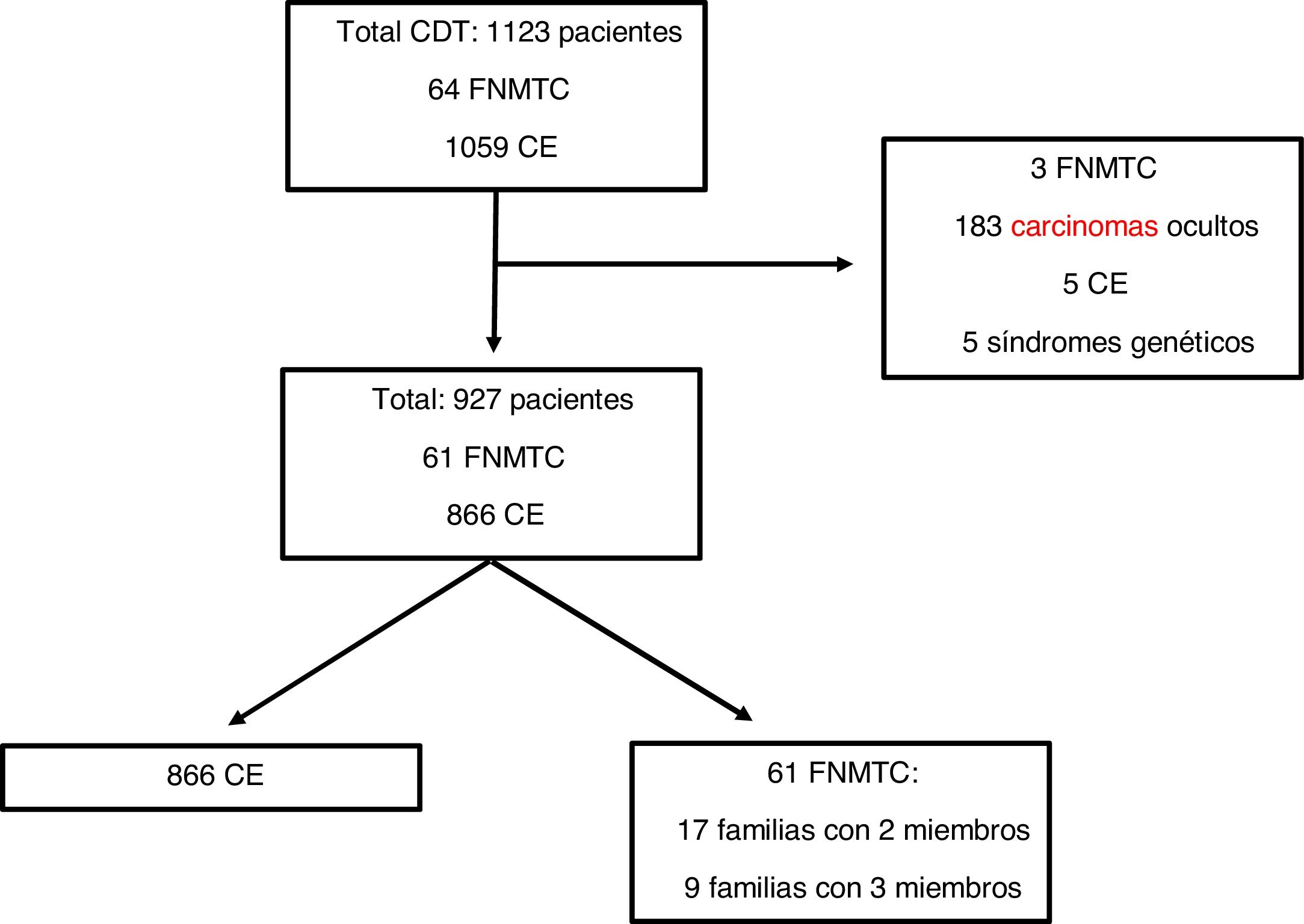
Material y métodosPacientes y métodosEl diagrama seguido con los pacientes incluidos y excluidos en el análisis se encuentra reflejado en la figura 1. Se evaluó a 1.123 pacientes de forma retrospectiva incluidos en la cohorte hospitalaria de CT de Navarra, diagnosticados y tratados en nuestro centro desde 1990 hasta 2018, con seguimiento posterior en nuestras consultas de Endocrinología. Para el presente trabajo excluimos a los pacientes con CDT como hallazgo quirúrgico incidental (n=183), aquellos con síndromes genéticos asociados a CT (n=5), 5 CE por falta de datos y 3 clasificados como FNMTC pero con ausencia de seguimiento de sus familiares en nuestro centro. Tras ello, la población final sobre la que se ha realizado el análisis ha sido de 927 pacientes, 866 con CE y 61 con FNMTC.

Los casos de FNMTC fueron definidos como 2o más familiares de primer grado con diagnóstico anatomopatológico de CT de origen folicular. Los familiares de primer grado incluidos eran los padres, hermanos y descendencia que estuviesen en seguimiento en nuestro centro. En esta línea, los 61 pacientes clasificados como FNMTC pertenecen a 26 grupos familiares diferentes, con 17 familias de 2 miembros afectados (34 pacientes) y las restantes 9 familias con 3 (27 pacientes).
Protocolo del estudioSe recogieron variables demográficas (edad, género), analíticas (título de anticuerpos antitiroglobulina, tiroglobulina basal, tiroglobulina estimulada, TSH, T4 libre), histopatológicas (tamaño, ganglios, anatomía patológica del tumor, multifocalidad, bilateralidad, invasión vascular, tamaño ganglios, extensión extratiroidea), tipo de tratamiento, dosis de I131 y número tratamientos con I131. También se analizaron los datos de captación en el rastreo corporal tras I131, el cálculo de riesgo de recurrencia, la estratificación dinámica (respuesta excelente, indeterminada, bioquímica incompleta, estructural incompleta) y la supervivencia libre de enfermedad y mortalidad por enfermedad tiroidea. La variable principal del trabajo ha sido la presencia de persistencia o recurrencia de la enfermedad al finalizar el estudio o en el momento del fallecimiento del paciente. Las características histológicas del tumor se definieron por patólogos con experiencia en tiroides.
Todos los pacientes fueron seguidos en nuestro centro al menos una vez al año. El seguimiento se ha realizado mediante una historia clínica, exploración física, determinación de tiroglobulina y anticuerpos antitiroglobulina, y ecografía cervical periódica. En función de la sospecha clínica, se realizó una determinación de tiroglobulina estimulada con rastreo corporal para detectar persistencia/recurrencia y se solicitaron otras pruebas de imagen (radiografía, tomografía computarizada, resonancia magnética y tomografía por emisión de positrones).
La ausencia de enfermedad se definió cuando la tiroglobulina estimulada era inferior a 1 ng/ml con negatividad de anticuerpos antitiroglobulina, rastreo corporal normal y con una ecografía cervical sin datos patológicos. Se unificaron las categorías de persistencia y recurrencia, definidas como la evidencia de enfermedad un año después del tratamiento inicial (tiroidectomía total más yodo radiactivo), mediante niveles de tiroglobulina detectables o prueba de imagen positiva confirmada con citología o cirugía. El estadio tumoral se evaluó según la 7.ª clasificación TNM de la edición del American Joint Comitee on Cancer (AJCC), realizada en el momento diagnóstico19.
Análisis estadísticoLas variables continuas están expresadas como media±desviación estándar y se utilizó el test de la t de Student para comparar estas variables. Las variables categóricas se expresan como números y porcentajes, y se comparan con la prueba de la chi al cuadrado. La comparación de la supervivencia libre de enfermedad entre los distintos grupos se realiza con las curvas de Kaplan-Meier y el test de Log-Rank. Se consideraron estadísticamente significativos aquellos valores p inferiores a 0,05. El estudio estadístico se llevó a cabo con el programa Stata versión 12 (Stata Statistical Software: College Station, TX: Stata Corp LP).
ResultadosLas características basales están reflejadas en la tabla 1. Se incluyó a un total de 927 pacientes, de los cuales 61 presentaban FNMTC (6,6%). Los restantes 866 (93,4%) se definieron como CE. El seguimiento medio ha sido de 9,7±6,5 años. La edad media de los pacientes es de 46,3±15,0 años, con una mayor prevalencia de mujeres (78%).
Descripción de la muestra y subgrupos. Comparación de características clinicopatológicas del CDT esporádico de tiroides frente al CDT familiar no medular
| Características basales de la muestra(927) | Cáncer esporádico (866) | Cáncer familiar(61) | Valor p | |
|---|---|---|---|---|
| Edad al diagnóstico (media±desviación estándar) | 46,3±15,0 | 46,5±15,1 | 44,6±14,1 | 0,347 |
| Sexo femenino (%) | 78 | 78 | 74 | 0,406 |
| Histología (%) | 0,022 | |||
| Papilar | 75 | 74 | 81 | |
| Folicular | 15 | 16 | 8 | |
| Cáncer de células de Hürthle | 8 | 8 | 4 | |
| Pobremente diferenciado | 2 | 2 | 7 | |
| Tamaño <2cm (%) | 41 | 40 | 60 | 0,003 |
| Multifocalidad (%) | 31 | 30 | 44 | 0,034 |
| Bilateralidad (%) | 18 | 17 | 26 | 0,071 |
| Invasión vascular (%) | 24 | 24 | 14 | 0,196 |
| Invasión linfática (%) | 40 | 41 | 32 | 0,333 |
| Enfermedad extratiroidea (%) | 14 | 14 | 14 | 0,900 |
| Afectación ganglionar (%) | 24 | 24 | 25 | 0,973 |
| Estadio TNM | ||||
| T1/T2/T3/T4% | 41/30/25/4 | 40/31/25/4 | 61/11/26/2 | 0,003 |
| N0/N1a/N1b % | 75/11/14 | 76/10/14 | 75/12/12 | 0,883 |
| M (%) | 5 | 5 | 5 | 0,953 |
| Estadio TNM | ||||
| I/II/III/IV | 64/16/13/7 | 64/17/12/7 | 75/2/16/7 | 0,028 |
| Tiroidectomía (%) | 99 | 99 | 96 | 0,068 |
| Linfadectomía (%) | 12 | 12 | 9 | 0,448 |
| Radioyodo (%) | 96 | 96 | 91 | 0,111 |
| Radioyodo dosis mCI(media±desviación estándar) | 145±135 | 145±136 | 140±124 | 0,787 |
| Persistencia / recurrencia | 13,8 | 13,7 | 15,8 | 0,656 |
| Libre de enfermedad | 78,7 | 78,7 | 77,6 | 0,929 |
| Mortalidad por CDT | 2,6 | 2,5 | 3,5 | 0,666 |
| Tiempo seguimiento (años) | 9,7±6,5 | 9,7±6,5 | 10,7±7,1 | 0,331 |
En la tabla 1 se comparan las características de los pacientes con FNMTC y CE. No se detectaron diferencias significativas respecto a la edad del diagnóstico (media 44,6±14,1 años vs. 46,5±15,1 años; p=0,347) o sexo femenino (74% vs. 78%; p=0,406). Se observa mayor presentación multifocal en el grupo de FNMTC (44% vs. 30%; p=0,034). La variante histológica predominante en ambos grupos ha sido el subtipo papilar, aunque este porcentaje estaba representado en mayor proporción en los FNMTC (81% vs. 74%; p=0,022). El tamaño tumoral del 60% de los pacientes con FNMTC ha sido inferior a 2cm, en contraposición al 40% de los CE (p=0,003), condicionando un estadificación TNM significativamente menor (p=0,003). No encontramos diferencias respecto a presentación bilateral de la enfermedad (26% vs. 17%; p=0,071), invasión linfática (32% vs. 41%; p=0,333), vascular (14% vs. 24%; p=0,196) y enfermedad extratiroidea (14% vs. 14%; p=0,900).
No se detectaron diferencias significativas en el abordaje terapéutico inicial. Se realizó tiroidectomía total en uno o 2tiempos en el 96% de los FNMTC frente al 99% de los CE (p=0,068). Se precisó linfadenectomía cervical en el 9 y el,12% de los pacientes, respectivamente (p=0,448). Posteriormente, se administró I131 en el 91% de los FNMTC y 96% de los CE (p=0,111), con una dosis acumulada comparable de 140±124 mCi en FNMTC y 145±136 mCi en CE (p=0,787).
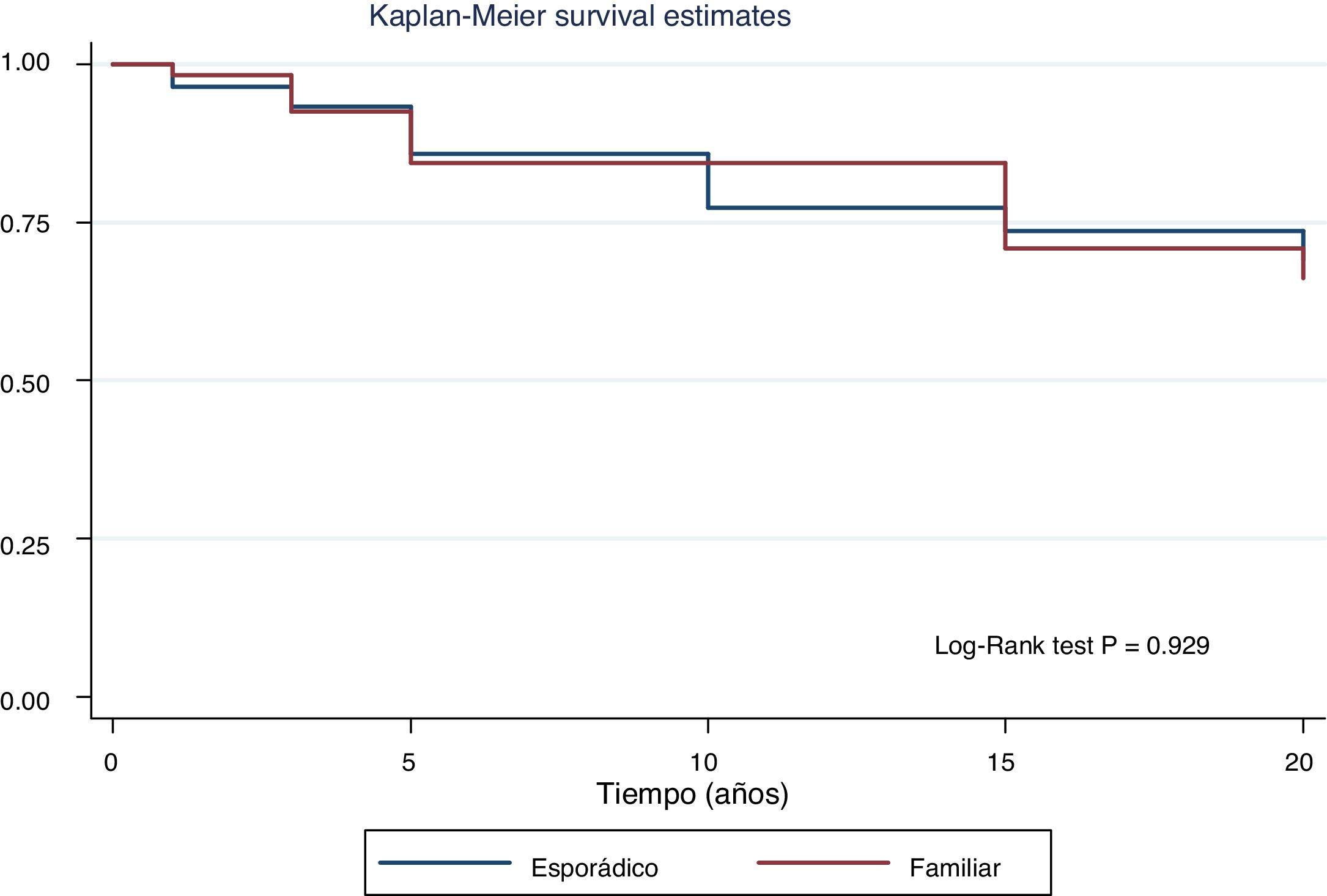
En la figura 2 observamos las curvas de supervivencia libre de enfermedad de ambos grupos. La supervivencia libre de enfermedad al fin del seguimiento ha sido similar (77,6% vs. 78,7%; p=0,929). Tampoco se observaron diferencias en la tasa de persistencia/recurrencia en el FNMTC frente al CE (15,8% vs. 13,7%; p=0,656) ni en la mortalidad derivada del carcinoma, que se situaba en un 3,5% del FNMTC frente al 2,5% en el CE (p=0,331).

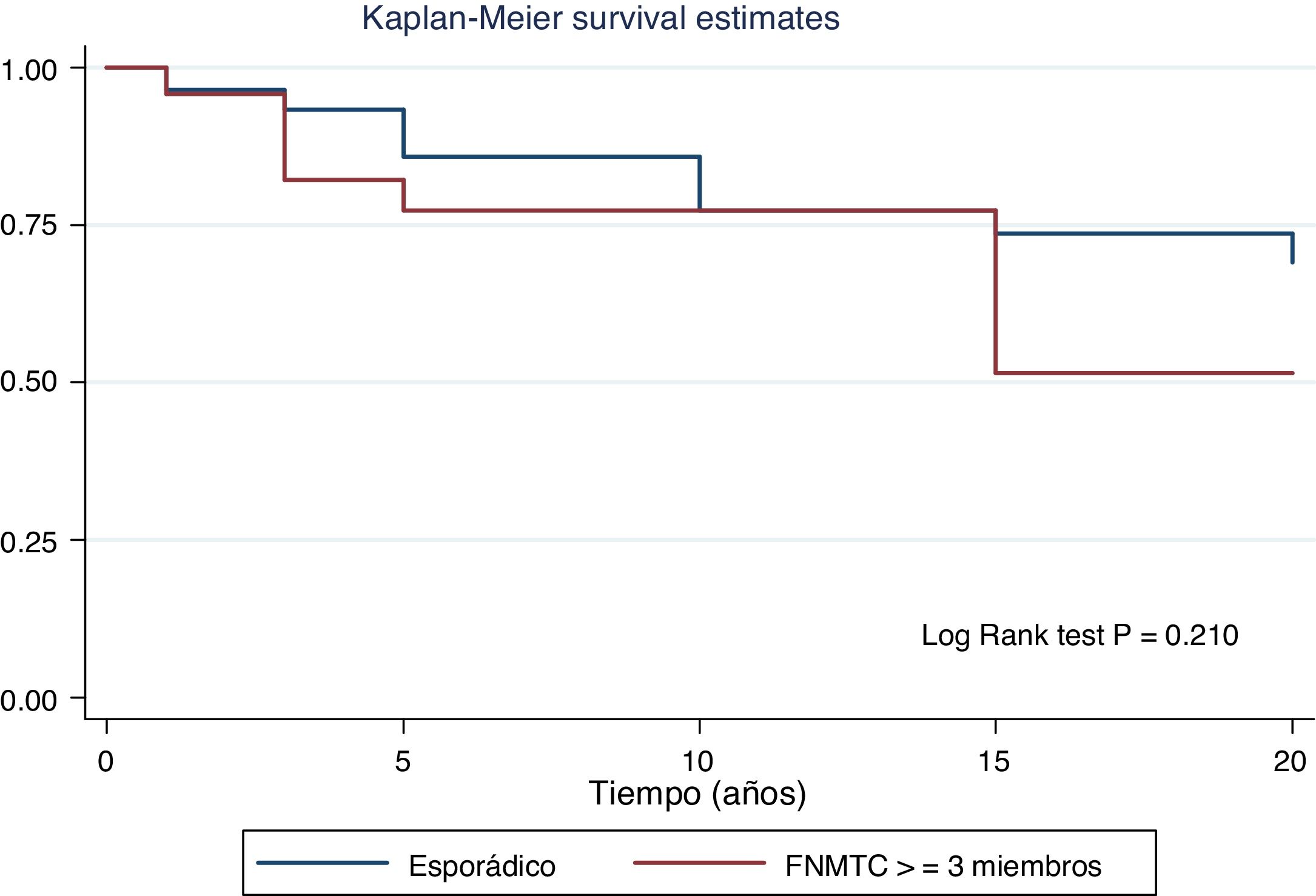
En la tabla 2 se detalla el subanálisis con las diferencias encontradas al comparar a los FNMTC en familias de 2y 3miembros afectados frente al CE. Se observó que en el grupo de 3o más familiares los tumores eran de menor tamaño (15,6±14,3mm vs. 26,5±16,9mm; p=0,002), con mayor multifocalidad (50% vs. 30%; p=0,040), bilateralidad (42% vs. 17%; p=0,002), mayor proporción de hombres (42% vs. 22%; p=0,020), siendo más frecuente el subtipo papilar (96% vs. 74%; p=0,049). Además, en el FNMTC de 3 miembros existe un menor número de intervenciones quirúrgicas mediante tiroidectomía (92% vs. 99%; p=0,001) y tratamientos con radioyodo (83% vs. 96%; p=0,004). En términos de pronóstico, la incidencia de persistencia/recurrencia, supervivencia libre de enfermedad (fig. 3) y mortalidad derivada del carcinoma han sido comparables en ambos grupos.
Comparación de características clinicopatológicas del CDT esporádico frente a FNMTC con familias de 2 miembros y frente a las de ≥ 3 miembros
| CDT esporádico(866) | FNMTC2 miembros (34) | Valor p | FNMTC≥ 3 miembros (27) | Valor p | |
|---|---|---|---|---|---|
| Edad al diagnóstico (media±desviación estándar) | 46,5±15,1 | 46,6±14,9 | 0,843 | 41,4±14,3 | 0,094 |
| Sexo femenino (%) | 78 | 84 | 0,374 | 58 | 0,020 |
| Histología (%) | |||||
| Papilar | 74 | 70 | 0,056 | 96 | 0,049 |
| Folicular | 16 | 15 | 0 | ||
| Cáncer de células de Hürthle | 8 | 6 | 0 | ||
| Pobremente diferenciado | 2 | 9 | 4 | ||
| Tamaño <2cm (%) | 40 | 58 | 0,200 | 75 | 0,005 |
| Multifocalidad (%) | 30 | 39 | 0,271 | 50 | 0,040 |
| Bilateralidad (%) | 17 | 15 | 0,788 | 42 | 0,002 |
| Invasión vascular (%) | 24 | 18 | 0,529 | 8 | 0,200 |
| Invasión linfática (%) | 41 | 37 | 0,733 | 26 | 0,279 |
| ETE (%) | 14 | 16 | 0,755 | 13 | 0,867 |
| Afectación ganglionar (%) | 24 | 21 | 0,679 | 29 | 0,589 |
| Estadio TNM | |||||
| T1/T2/T3/T4% | 40/31/25/4 | 50/10/34/6 | 0,072 | 75/17/4/4 | 0,005 |
| N0/N1a/N1b % | 76/10/14 | 79/6/15 | 0,711 | 71/21/8 | 0,233 |
| M % | 5 | 6 | 0,803 | 4 | 0,839 |
| Estadio (I, II, III, IV) | 64 / 17 / 12 / 7 | 60/6/25/9 | 0,124 | 92/0/0/8 | 0,020 |
| Tiroidectomía (%) | 99 | 100 | 0,579 | 92 | 0,001 |
| Linfadectomía (%) | 12 | 9 | 0,597 | 8 | 0,572 |
| Radioyodo (%) | 96 | 97 | 0,748 | 83 | 0,004 |
| Radioyodo dosis mCI(media±desviación estándar) | 145,7±136,0 | 157,2±113,5 | 0,633 | 118,1±136,6 | 0,326 |
| Persistencia / recurrencia | 13,7 | 12,1 | 0,788 | 20,8 | 0,324 |
| Libre de enfermedad | 78,7 | 81,8 | 0,422 | 70,8 | 0,210 |
| Mortalidad por CDT | 2,5 | 6,1 | 0,287 | 0 | 0,465 |
| Tiempo seguimiento (años) | 9,7±6,5 | 12,6±7,3 | 0,013 | 8,1±5,9 | 0,248 |
Datos presentados en negrita, aquellos estadísticamente significativos (p < 0,05).

De los 61 FNMTC, 34 (56%) presentan una relación paterno filial y 27 de fraternidad. Analizando los 34 casos de padres e hijos, 15 pacientes pertenecieron a la primera generación y 19 eran descendientes, por lo tanto, de segunda generación. Al comparar ambas generaciones en la tabla 3, se aprecia que la edad al diagnóstico es 23,1 años menor en los miembros de la segunda generación respecto a la primera (34,8±12,5 años vs. 57,9±12,8 años; p=0,001). A pesar de ello, el tamaño, las características anatomopatológicas y el pronóstico resultaron similares (p> 0,05).
Comparación de las características clinicopatológicas y pronósticas entre primera y segunda generación
| 1.ª generación(15) | 2.ª generación(19) | Valor p | |
|---|---|---|---|
| Edad al diagnóstico (media±desviación estándar) | 57,9±12,8 | 34,8±12,5 | 0,001 |
| Sexo femenino (%) | 80 | 73 | 0,666 |
| Histología papilar (%) | 73 | 89 | 0,235 |
| Tamaño en mm(media±desviación estándar) | 23,0±16,8 | 21,5±19,3 | 0,220 |
| Multifocalidad (%) | 40 | 36 | 0,851 |
| Bilateralidad (%) | 40 | 36 | 0,851 |
| Afectación ganglionar (%) | 20 | 36 | 0,285 |
| Metástasis | 13 | 5 | 0,410 |
| Tasa de recurrencia | 33 | 15 | 0,231 |
| Libre de enfermedad al fin del seguimiento | 60 | 84 | 0,112 |
Datos presentados en negrita, aquellos estadísticamente significativos (p < 0,05).
El FNMTC ha sido definido como una entidad diferenciada que representa entre el 5 y el 10% de todos los CDT. En este estudio encontramos que la prevalencia de FNMTC es del 6,6% (61/927), situándose en el límite inferior del porcentaje que describen otras series8,13,20, alejada del 9,6% de otros registros, como los de Corea del Sur o Norteamérica20,21. La falta de marcadores clínicos y genéticos en el CDT hace al FNMTC indistinguible del CE. Debido a la alta incidencia y supervivencia del CDT, se ha estimado que el 31-38% de los carcinomas con 2familiares afectados y el 94% con 3miembros son realmente casos de FNMTC y no CE8. Esto en nuestra muestra podría suponer que 34 pacientes de los 61 serían verdaderos casos con agregación familiar.
Algunos estudios han descrito un comportamiento más agresivo del FNMTC y peor pronóstico respecto al esporádico, mientras que otros obtienen resultados contrarios. La característica más repetida en diferentes estudios es una mayor multifocalidad y bilateralidad, en la línea de nuestra cohorte. Zhang et al.7 encuentran tasas de multifocalidad en el FNMTC frente al CE del 60% vs. 40% y bilateralidad del 44% vs. 29%, respectivamente (p<0,05). Cao et al.17, en su estudio de casos y controles, detectan en el grupo familiar una tasa de multifocalidad y bilateralidad del 54 y el 44%, respectivamente, frente al 39 y el 28% de los esporádicos (p <0,05).
El menor tamaño tumoral descrito por nuestro estudio es más controvertido según la bibliografía consultada. En nuestro caso, incluso implica menor estadificación TNM y tratamientos menos agresivos en el grupo de 3 miembros afectados. Lakis et al.16, al estudiar 78 FNMTC, observan una mayor proporción de tumores T1. Sin embargo, un metaanálisis realizado en 2015 que incluía 12 estudios con pacientes procedentes de Asia, Norteamérica y Europa22 no encontraba diferencias en lo que respecta al tamaño. En nuestra cohorte existe globalmente una proporción de tumores inferiores a 2 cm del 41%, muy por debajo del 75% descrito por Lakis et al.16. El mayor tamaño de los carcinomas de nuestra serie puede ser debido a la exclusión de los microcarcinomas detectados de forma incidental en cirugías benignas de tiroides. Por lo tanto, no se incluyen a aquellos CDT ocultos inferiores a un centímetro que son descubiertos en el análisis anatomopatológico de la pieza quirúrgica de manera inesperada23. Esto acentúa las diferencias entre el grupo familiar y esporádico, presentando el FNMTC una mayor proporción de carcinomas T1 (61%) frente al 40% de los CE (p=0,003).
Zhang et al.24 ponían de manifiesto que los FNMTC presentaban mayor enfermedad ganglionar (52,6% vs. 33,3%; p <0,05) y afectación extratiroidea (64,1% vs. 48,5%; p <0,05). En nuestra muestra no se detectaron diferencias significativas en estos 2aspectos.
Un número creciente de estudios ha demostrado una mayor agresividad del FNMTC, especialmente acentuada en el grupo de 3 miembros afectados. Alsanea et al.25 detectaron en FNMTC mayores tasas de recurrencia (44% vs. 17%; p <0,05) junto a una menor supervivencia libre de enfermedad respecto al CE. McDonald et al.26 detectaron que a los grupos con 3o más familiares afectados de CDT se les realizaba más reintervenciones (p=0,05) o requerían dosis adicionales de radioyodo (p=0,03), presentaban más metástasis a distancia (p=0,003) y muertes (p=0,01).
Según nuestros datos, a pesar de la mayor multifocalidad (p=0,040) y bilateralidad (p=0,002), que podría ser un marcador temprano de agresividad del FNMTC, el pronóstico en forma de persistencia/recurrencia (p=0,324) y mortalidad por CDT (p=0,465) es equiparable, sin encontrar datos indicativos de una peor evolución. No se ha realizado cribado que podría justificar tales resultados, pero una mayor conciencia sobre el CDT en el grupo familiar podría explicar un diagnóstico en una fase más temprana y, por tanto, un tratamiento menos agresivo con un pronóstico equiparable.
La mayoría de los estudios demuestran que la edad de presentación del FNMTC es menor respecto al esporádico7. Moses y Weng20 describen que la edad al diagnóstico se adelanta de media 5 años. Sin embargo, este dato podría estar afectado por el fenómeno de anticipación de la segunda generación publicado por Capezzone et al.2 en 2008. En su cohorte italiana, demostró que el diagnóstico del CDT en la segunda generación se anticipa respecto a sus progenitores. Esto también ha sido confirmado posteriormente por el estudio de Zhou et al.27, entre otros. En nuestra muestra, la edad de presentación del FNMTC es 23,1 años menor en la segunda generación. Si esta anticipación fuera únicamente reflejo de un diagnóstico precoz, cabría esperar otras diferencias, como un menor tamaño tumoral o menor agresividad. Sin embargo, el resto de las variables han sido equiparables, lo que apoya la existencia de este fenómeno genético que se muestra en otros procesos tanto benignos como malignos.
Encontramos en el grupo de 3 miembros afectados una mayor proporción de varones. Este hallazgo también lo describe Lakis et al.16 con una cohorte de 78 FNMTC, así como Cao et al.17 en un estudio de casos y controles con 372 casos. Esto es congruente con la herencia autosómica dominante que se sospecha de esta entidad. Ante este factor genético, la predominancia femenina del CE desaparece en el FNMTC, habiendo un mayor número de hombres. El CDT, en concreto el subtipo papilar, muestra un elevado riesgo cuando un familiar de primer grado o gemelo es diagnosticado, siendo uno de los tumores que presenta mayor ratio de incidencia estandarizada (RIE), con valores de 3,21 y 6,24, respectivamente28. Probablemente se deba a la presencia de un grupo de genes con herencia autosómica dominante y penetrancia incompleta, siendo las madres las que lo transmiten con mayor susceptibilidad (RIE 4,32)29. La mayor incidencia de CDT papilar de manera global en mujeres sugiere la existencia de un papel favorecedor de la progresión o transformación maligna a nivel tiroideo secundario a la estimulación estrogénica30. En el inicio de la pubertad, la incidencia del CDT aumenta solo en el género femenino, volviendo a disminuir tras la menopausia, posiblemente por el efecto de promoción de crecimiento mediado por receptores de estrógeno ligados a membrana31.
En lo que respecta al mecanismo de transmisión, Tavarelli et al.32, en un estudio retrospectivo con 151 individuos de 74 familias, detectan que lo más frecuente es que ocurra entre hermanos (62,2%), seguido de la herencia materna (26,5%) y en último lugar la paterna (7,3%). En nuestra muestra, 27 pacientes (44%) presentaban una relación de fraternidad, seguido de 34 (56%) pacientes con relación paterno filial, siendo en 3 casos la relación paternal y 12 maternal.
Nuestros resultados son consistentes con estudios previos, que concluyen que las diferencias entre el CE y el FNMTC son debidas al grupo de familiares con 3 miembros afectados. Esto indica que las familias con 3 o más miembros con FNMTC constituyen los verdaderos casos familiares y pacientes con 2 familiares puedan representar una agrupación de 2casos esporádicos.
La principal fortaleza del estudio es que recoge a todos los pacientes diagnosticados de CDT en un área geográfica durante un periodo relativamente largo, lo que refleja la población con la que trabajamos diariamente. No se ha realizado cribado diagnóstico y el tratamiento realizado fue equiparable en ambos grupos, lo que da validez a los resultados pronósticos.
También presenta varias limitaciones. En primer lugar, dada la baja prevalencia de dicha patología, el tamaño muestral de la cohorte de FNMTC es modesto. Los datos provienen de una única comunidad autónoma y han sido analizados de manera retrospectiva, con la posible existencia de sesgos para universalizar las conclusiones. Finalmente, dada la ausencia de marcadores y genes que confirmen específicamente que se trate de un FNMTC, un porcentaje de los casos con 2miembros diagnosticados de CDT clasificado como familiar en realidad serán 2CE que coincidan en una misma familia (debido a la relativa alta incidencia del CDT). Sin embargo, esto da más valor a las diferencias encontradas, puesto que su inclusión solo podría atenuar la agresividad demostrada.
En conclusión, el FNMTC se ha constituido como una entidad diferenciada dentro del CDT. Las familias con 3 o más miembros de FNMTC constituyen los verdaderos casos familiares y se presentan con tumores más pequeños, multifocales, bilaterales, mayor representación del subtipo papilar y de hombres. Sin embargo, en el presente estudio no se han encontrado diferencias pronósticas ni de afectación ganglionar, por lo que los datos no son suficientes para justificar un tratamiento más agresivo. Por ello, el tratamiento inicial debería plantearse de forma similar al CE, en función del riesgo y la estadificación TNM inicial. En lo que respecta al abordaje quirúrgico, se debería tener en cuenta la mayor bilateralidad. No precisan un seguimiento más intensivo, ya que los datos de recurrencia y persistencia son similares, con una esperanza de vida que no se ve condicionada por el propio tumor. A pesar de la agregación y el aumento de riesgo de hasta 3veces de presentar CDT en los familiares con más de 3miembros afectados, no existe indicación actualmente de realizar cribado para su detección precoz debido al buen pronóstico. Posiblemente, y según los datos expuestos, sería razonable estudiar a los familiares del subgrupo con 3o más miembros diagnosticados de CDT. En estas familias de alto riesgo, debería tenerse en cuenta el fenómeno de anticipación en la segunda generación para definir la edad a la que llevar a cabo las pruebas.
Conflicto de interesesLos autores no declaran ningún conflicto de intereses.
Los autores agradecen a todos los componentes del comité multidisciplinar de cáncer de tiroides del Complejo Hospitalario de Navarra, compuestos por los servicios de Endocrinología y Nutrición, Cirugía General, Anatomía Patológica, Radiodiagnóstico y Oncología Médica.
