




El carcinoma diferenciado de tiroides familiar se define como la presencia de cáncer de tiroides no medular en dos o más familiares de primer grado, en ausencia de otros factores predisponentes. Representa hasta el 9% de los cánceres diferenciados de tiroides, y solo una minoría aparece en el seno de síndromes hereditarios bien conocidos que asocian el cáncer de tiroides, entre otras múltiples manifestaciones clínicas. Sin embargo, en más del 95% de los casos, el cáncer de tiroides aparece de manera aislada, y sus causas genéticas aún están por dilucidar. En esta revisión resumimos el estado actual del conocimiento de las bases genéticas de esta patología, así como sus características clínicas. La comprensión de los mecanismos genéticos implicados ayudaría a esclarecer las vías metabólicas involucradas, con la consiguiente potencial aplicación terapéutica. Además, permitiría ofrecer consejo genético y focalizar nuestros esfuerzos en los pacientes susceptibles de padecer la patología.
Familial non-medullary thyroid cancer is defined as the presence of non-medullary thyroid cancer in two or more first-degree relatives, in the absence of other predisposing factors. It represents up to 9% of differentiated thyroid cancers, and only a minority appears in well-known hereditary syndromes that associate thyroid cancer among many other clinical manifestations. However, in more than 95% of cases, thyroid cancer appears isolated, and its genetic causes have yet to be elucidated. We review here the current knowledge of the genetic basis of this pathology, as well as its clinical characteristics. Understanding the genetic mechanisms implied would help to comprehend the metabolic pathways involved, with the consequent potential therapeutic application. In addition, it would allow genetic counseling and to focus our efforts on patients at risk of developing this disorder.
El cáncer de tiroides es la neoplasia endocrina más común, representando actualmente el quinto cáncer más frecuente en mujeres en EE. UU.1 El 95% de las neoplasias de tiroides son carcinomas diferenciados de tiroides (CDT), derivados de las células foliculares: los carcinomas papilares (aproximadamente el 80% de los casos), los carcinomas foliculares (15%) y otros tipos de cáncer menos frecuentes (< 5%). El 5% de neoplasias de tiroides restantes derivan de las células C o parafoliculares, productoras de calcitonina, ocasionando los carcinomas medulares de tiroides (CMT)2.
Existen familias en las que concurren varios pacientes afectos de cáncer de tiroides. Su abordaje asistencial integral, que posiblemente incluye asesoramiento genético, requiere conocer las bases genéticas de la enfermedad. En el caso del CMT familiar (hasta un 25% de los CMT), se sabe que el protooncogén RET es el principal protagonista implicado3. La presencia de mutaciones germinales en RET se asocia con el CMT hereditario o la neoplasia endocrina múltiple tipo 2, con un patrón de herencia autosómica dominante (HAD). En los últimos años, se ha avanzado mucho en el conocimiento genético de esta patología, e incluso se ha podido establecer una estrecha correlación genotipo-fenotipo4. Sin embargo, en esta revisión nos centraremos en el carcinoma familiar no medular, cuya base hereditaria conocemos mucho menos.
El primer paso es definir esta entidad: el carcinoma diferenciado (o no medular) de tiroides familiar, también conocido como FNMTC (familial non-medullary thyroid cancer), se define por la presencia de esta neoplasia en dos o más familiares de primer grado, siempre y cuando no haya otros factores predisponentes como radiación o deficiencia de yodo5.
En segundo lugar, debemos saber que la prevalencia de FNMTC no es desdeñable: se estima que hasta el 9% de los carcinomas no medulares de tiroides presentan un componente hereditario5. Por tanto, es lo suficientemente frecuente como para que identifiquemos algunos casos en nuestro ámbito clínico; y al mismo tiempo es tan infrecuente como para que debamos aunar esfuerzos multicéntricos con el fin de reunir una casuística que permita avanzar en su investigación.
En tercer lugar, una vez identificados los posibles FNMTC, debemos distinguir entre los casos sindrómicos y no sindrómicos. Es decir, ¿el FNMTC se presenta de manera aislada, o bien es parte del espectro fenotípico de un síndrome? En el 95% de los casos estaremos ante un FNMTC no sindrómico. No obstante, alrededor del 5% de los casos de FNMTC aparece en el seno de síndromes genéticos bien caracterizados6, que debemos conocer por sus posibles repercusiones sistémicas, que exigen un abordaje multidisciplinar.El objetivo de la presente revisión es resumir el conocimiento que existe hasta la actualidad sobre las bases genéticas del FNMTC, centrándonos en el no sindrómico. Para ello, se procedió a la búsqueda bibliográfica en la base de datos MEDLINE, y se seleccionaron las revisiones o artículos originales más relevantes publicados durante los últimos 20 años (1999-2020), en relación con este tema. Las palabras clave empleadas en la búsqueda fueron «thyroid cancer», «familial», «genetics», «germline mutations» y «non-medullary».
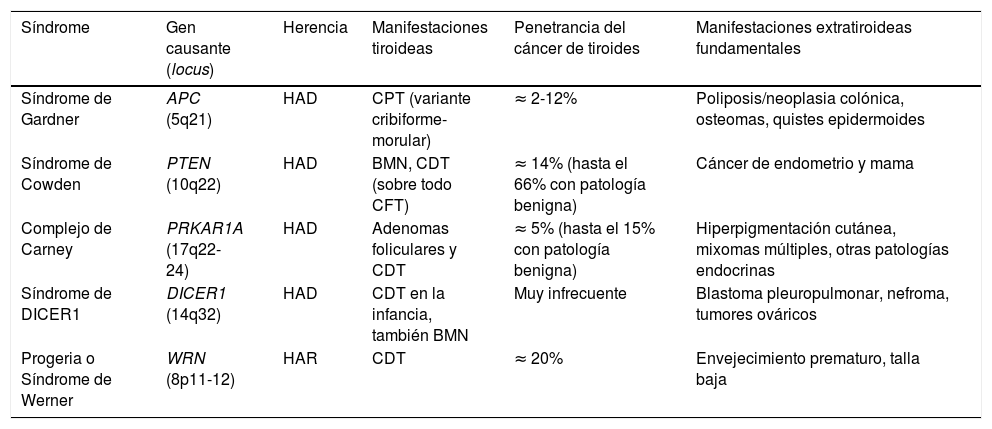
Carcinoma no medular de tiroides familiar sindrómicoHoy en día se reconocen los siguientes cinco síndromes que cursan con FNMTC, cuyos genes involucrados ya han sido descritos y que repasaremos a continuación someramente (tabla 1). En otros síndromes como el McCune-Albright, el Peutz-Jeghers o el de ataxia-telangiectasia, se ha sugerido asimismo una predisposición al cáncer diferenciado de tiroides no medular, pero ésta no se encuentra claramente establecida.
Síndromes hereditarios asociados con FNMTC
| Síndrome | Gen causante (locus) | Herencia | Manifestaciones tiroideas | Penetrancia del cáncer de tiroides | Manifestaciones extratiroideas fundamentales |
|---|---|---|---|---|---|
| Síndrome de Gardner | APC (5q21) | HAD | CPT (variante cribiforme-morular) | ≈ 2-12% | Poliposis/neoplasia colónica, osteomas, quistes epidermoides |
| Síndrome de Cowden | PTEN (10q22) | HAD | BMN, CDT (sobre todo CFT) | ≈ 14% (hasta el 66% con patología benigna) | Cáncer de endometrio y mama |
| Complejo de Carney | PRKAR1A (17q22-24) | HAD | Adenomas foliculares y CDT | ≈ 5% (hasta el 15% con patología benigna) | Hiperpigmentación cutánea, mixomas múltiples, otras patologías endocrinas |
| Síndrome de DICER1 | DICER1 (14q32) | HAD | CDT en la infancia, también BMN | Muy infrecuente | Blastoma pleuropulmonar, nefroma, tumores ováricos |
| Progeria o Síndrome de Werner | WRN (8p11-12) | HAR | CDT | ≈ 20% | Envejecimiento prematuro, talla baja |
BMN: bocio multinodular; CDT: carcinomas diferenciados de tiroides; CFT: carcinomas foliculares de tiroides; CPT: carcinomas papilares de tiroides; HAD: herencia autosómica dominante; HAR: herencia autosómica recesiva.
El síndrome de Gardner es un síndrome de HAD, causado por mutaciones germinales en el gen supresor tumoral APC (cromosoma 5q21). Se trata de una variante de la poliposis adenomatosa familiar, en la que además concurren manifestaciones extracolónicas. Clínicamente cursa con poliposis/neoplasia colónica, osteomas, quistes epidermoides y tumores dermoides, entre otros. Entre un 2-12% de los pacientes pueden desarrollar CDT, habitualmente a edades jóvenes, y como primera manifestación del síndrome hasta en un tercio de los casos. Concretamente se asocia a la variante cribiforme-morular del carcinoma papilar, por lo que ante cualquier paciente con dicha histología debemos descartar que no se trate de la manifestación inicial de un síndrome de Gardner7.
Síndrome de Cowden [MIM: 158350]Éste es un síndrome de HAD asociado a mutaciones inactivadoras en línea germinal en el gen supresor PTEN (cromosoma 10q22). Los pacientes padecen múltiples hamartomas y mayor riesgo de cáncer de endometrio y mama, y hasta 2/3 de los pacientes tienen patología tiroidea, principalmente bocio multinodular (BMN) y CDT (este último en un 3-14% de los casos, y fundamentalmente de estirpe folicular)7. En pacientes en los que no se hallan mutaciones en PTEN (hasta un 15%), deberían considerarse otros genes más infrecuentemente asociados al síndrome de Cowden, tales como SDHB-D, la hipermetilación del promotor de KLLN8; PIK3CA y AKT19; RASAL110 y SEC23B11.
Complejo de Carney [MIM: 160980]Se trata de un síndrome de HAD debido a mutaciones germinales en el gen supresor PRKAR1A (cromosoma 17q22-24), que provoca hiperpigmentación cutánea (a destacar léntigos y nevus azules), mixomas en múltiples localizaciones y patología endocrina hipofisaria, suprarrenal, gonadal, y/o tiroidea. Hasta un 15% de los afectados pueden presentar adenomas y más raramente carcinomas de tiroides. No ha de confundirse con el término «tríada de Carney» (asociación de tumor del estroma gastrointestinal [GIST] + condroma pulmonar + paraganglioma)7.
Síndrome de DICER1 [MIM: 606241]De HAD, y de descripción más reciente, se relaciona con mutaciones germinales en DICER1 (cromosoma 14q32), que codifica una ribonucleasa, predisponiendo a múltiples tumores en la infancia como el blastoma pleuropulmonar, así como al bocio multinodular (BMN) y CDT. Es un gen a tener en cuenta fundamentalmente en las neoplasias tiroideas en edad pediátrica12.
Progeria o síndrome de Werner [MIM: 277700]Es el único de estos síndromes de herencia autosómica recesiva (HAR), originado por mutaciones en el gen WRN (cromosoma 8p11–12), que codifica una helicasa implicada en el mantenimiento de los telómeros. Sus características clínicas incluyen talla baja, múltiples neoplasias (entre ellas de tiroides, hasta en un 20% de los casos), y lo más distintivo: un envejecimiento prematuro. Es muy infrecuente fuera de Japón7.
Carcinoma no medular de tiroides familiar, no sindrómicoLa mayoría de nuestros pacientes con cáncer de tiroides presentan subtipos histológicos no medulares (mayoritariamente carcinomas papilares o foliculares), que en aproximadamente el 9% presentan antecedentes familiares. Excluyendo los pocos que aparecen en el seno de los síndromes comentados, el resto son no sindrómicos, y su genética está aún por dilucidar. Seguidamente, revisaremos las características clínicas de los FNMTC no sindrómicos, para posteriormente centrarnos en los genes de susceptibilidad descritos hasta la fecha.
Características clínicasHace más de 10 años que el FNMTC es reconocido como una enfermedad con entidad propia, que representa aproximadamente el 3,2-6,2% de todos los cánceres de tiroides5. Sin embargo, y dada la alta prevalencia e incidencia de cáncer de tiroides en la población general (se diagnostican 15,8 nuevos casos/100.000 habitantes/año13), surge la duda de si en realidad la presencia de dos o más casos de cáncer no medular de tiroides en una misma familia no podrían deberse al azar. Tras un complejo análisis estadístico, Charkes14 concluyó que alrededor del 62% de las familias con dos casos pueden tratarse de fenocopias (dos casos esporádicos asociados por azar). En cambio, si hay tres miembros afectos, la probabilidad de que sea realmente hereditario es del 96%.
Se ha observado que el FNMTC presenta HAD, con expresividad variable y penetrancia incompleta. La mayoría de los FNMTC son carcinomas papilares (≈ 90%), seguidos de foliculares, al igual que en los carcinomas no medulares esporádicos. Dentro de la misma familia pueden coexistir diferentes variantes histológicas. Respecto a alteraciones moleculares a nivel somático, al comparar las muestras de tiroidectomía/punción aspiración con aguja fina (PAAF) de los pacientes con FNMTC con las de los pacientes con CDT sin antecedentes familiares, no se observaron diferencias significativas en cuanto a frecuencia, ni tipo de mutaciones somáticas, con presencia de la mutación BRAF Val600Glu o mutaciones en NRAS o reordenamientos RET/PTC en un porcentaje muy similar de casos6. Esto va en favor de que las causantes de la enfermedad sean mutaciones en línea germinal, sin que las mutaciones somáticas jueguen un papel crucial en la tumorogénesis.
Globalmente, las características clínicas son similares, con una predominancia femenina en algunos estudios (proporción 2-3/1). Como rasgos diferenciales, el FNMTC suele presentarse con una edad menor al diagnóstico, en torno a los 43 años –versus los 48 años del CDT esporádico–6. Incluso estudios recientes sugieren que la segunda generación de afectos por el FNMTC podría presentar un «fenómeno de anticipación», con un diagnóstico a edad más temprana y una enfermedad más agresiva que en sus antecesores, sin descartar que en parte pueda ser debido a una más pronta observación de estas personas debido a sus antecedentes familiares. También habría mayor proporción de varones afectos15,16.
Además, en el FNMTC se ha descrito una mayor agresividad tumoral, según un metaanálisis de 2015 que incluyó 12 estudios con resultados bastante dispares17. Concretamente, se observó mayor presencia de adenopatías y extensión extratiroidea, mayor tasa de multifocalidad y de recurrencia, y menor supervivencia libre de progresión, especialmente en los estudios que incluyeron familias con al menos tres miembros afectos; las diferencias en las familias con sólo dos miembros afectos eran menos claras. En la misma línea, Ya-Bing Zhang et al.18 describen mayor agresividad en familias con tres o más miembros con FNMTC, sugiriendo que posiblemente los estudios deberían focalizarse en estas familias con mayor número de pacientes afectados.
Se ha investigado la utilidad de llevar a cabo un cribado de CDT en personas con familiares que sufren esta enfermedad19. En este estudio prospectivo de cinco años, el CDT fue detectado por cribado en un 4,6% de los individuos en riesgo de familias de dos miembros afectos, mientras que en las familias de ≥ 3 miembros afectos, el cribado detectó un 22,7% de CDT, concluyéndose que esta búsqueda activa debería ser considerada en estas últimas. Es decir, cuantas más personas afectas, mayor es el riesgo de que más miembros de la familia padezcan la patología, y estaría más indicado plantear una búsqueda activa20. También documentaron que los FNMTC detectados por cribado tenían menor tamaño tumoral, menor tasa de adenopatías y un tratamiento quirúrgico y de radioyodo menos agresivo que los casos índice19. Pese a todo, en las últimas guías de manejo del CDT de la American Thyroid Association (ATA)21, no se posicionan en favor o en contra del cribado familiar, ya que la reducción de la morbi-mortalidad no está demostrada. En realidad, esta demostración es poco factible en una enfermedad con una supervivencia a los 10 años del 95-97%. Triponez et al.22 compararon la supervivencia de pacientes con FNMTC con la de sus familiares no afectos y con población general, y observaron una supervivencia más corta en los pacientes cuyas familias eran de tres o más miembros afectos. Sin embargo, no compararon con CDT esporádicos.
En cualquier caso, la mayoría de las publicaciones reconocen que la mayor agresividad del FNMTC sigue siendo hoy un tema de debate debido a la heterogeneidad de los estudios, fundamentalmente retrospectivos. Esta controversia se extrapola al seguimiento y tratamiento de dichos tumores. Recientemente, se ha sugerido que el tratamiento quirúrgico inicial del FNMTC debería ser más agresivo, una vez más, en familias con ≥ tres miembros afectos23. De hecho, en las últimas guías de manejo del CDT21 se aboga por la tiroidectomía total y la terapia con radioyodo en pacientes con antecedentes familiares de CDT. Algún estudio sugiere además que, dado que estos pacientes podrían tener más adenopatías, habría que considerar la realización de linfadenectomía central junto a la tiroidectomía total24. Lo que no estaría indicado es la tiroidectomía total profiláctica, pues aún no hemos identificado las causas genéticas de la enfermedad que permitan detectar a los portadores, y además no existe una penetrancia completa. En cuanto a la PAAF, actualmente se recomienda en lesiones nodulares por encima de 1 cm25 (en las anteriores guías de la ATA de 2009, se recomendaba también en nódulos subcentimétricos). Se ha descrito que la PAAF en los pacientes con FNMTC podría tener más falsos negativos, dado que estos pacientes suelen tener una alta incidencia de enfermedad maligna multifocal, coexistente con nódulos benignos26.
En resumen, parece razonable considerar la presencia de un componente familiar como un factor de riesgo más a tener en cuenta en el manejo personalizado del cáncer de tiroides27, especialmente en familias con ≥ 3 miembros afectos, dada la alta probabilidad de que sea hereditario, y no por asociación casual; así como a la vista de los estudios que en este subgrupo han demostrado mayor agresividad tumoral y mayor beneficio de un cribado precoz.
Bases genéticasSi bien el FNMTC es una enfermedad relativamente poco prevalente, representa un esfuerzo de recursos significativo para el sistema nacional de salud, principalmente por el actual desconocimiento de sus determinantes hereditarios, y de sus posibles diferencias clínicas respecto a la enfermedad esporádica. Estas circunstancias dificultan su diagnóstico precoz (en estadios en los que el tratamiento es más resolutivo), y hace inviable el consejo genético que, además del impacto sanitario, es una demanda creciente de la sociedad actual. Conocer la mutación implicada en una neoplasia genética tendría como consecuencia directa permitir el estudio familiar, identificar a las personas portadoras de la mutación, concentrar en estas personas los esfuerzos diagnósticos y terapéuticos, y poder incidir precozmente en el curso de estas neoplasias. Asimismo, se evitaría tener que seguir clínicamente a todos los familiares no portadores. Por otra parte, la historia científica reciente nos señala que la identificación de genes causales de neoplasias hereditarias ha permitido un enorme avance en el conocimiento de la oncogénesis en los casos esporádicos (mucho más prevalentes), en la identificación de componentes y vías de señalización celulares implicadas, y en el diseño de moléculas capaces de modular estas vías y ejercer acciones terapéuticas específicas. Por todo, el conocimiento de las bases genéticas de una patología como el FNMTC resulta relevante por las implicaciones directas que puede tener en la práctica asistencial.
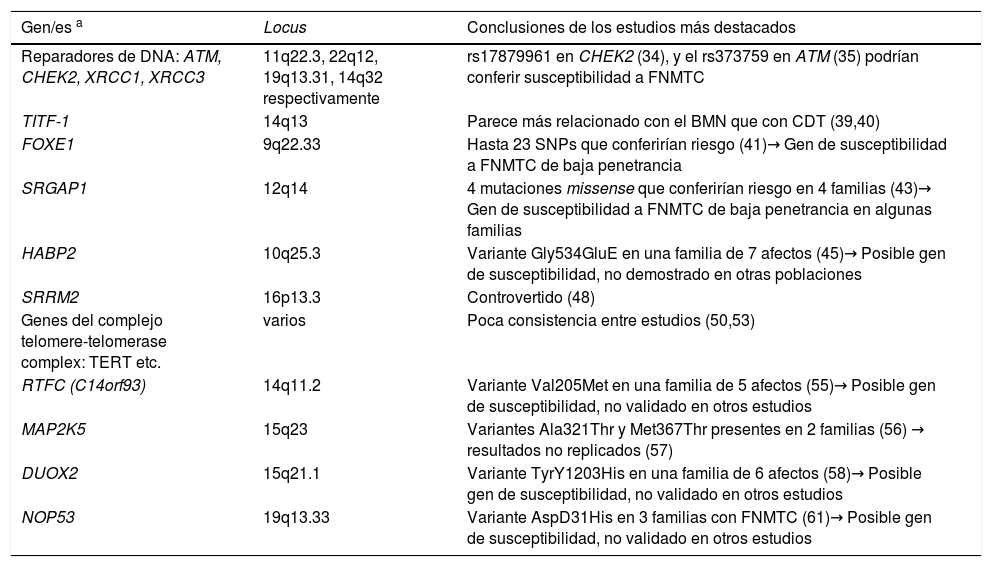
Se han empleado distintas estrategias de búsqueda de los genes implicados en esta patología, que detallamos a continuación. En la tabla 2 se resumen los principales genes candidatos identificados en la actualidad, y que describiremos posteriormente.
Genes/familias de genes descritos en el FNMTC no sindrómico
| Gen/es a | Locus | Conclusiones de los estudios más destacados |
|---|---|---|
| Reparadores de DNA: ATM, CHEK2, XRCC1, XRCC3 | 11q22.3, 22q12, 19q13.31, 14q32 respectivamente | rs17879961 en CHEK2 (34), y el rs373759 en ATM (35) podrían conferir susceptibilidad a FNMTC |
| TITF-1 | 14q13 | Parece más relacionado con el BMN que con CDT (39,40) |
| FOXE1 | 9q22.33 | Hasta 23 SNPs que conferirían riesgo (41)→ Gen de susceptibilidad a FNMTC de baja penetrancia |
| SRGAP1 | 12q14 | 4 mutaciones missense que conferirían riesgo en 4 familias (43)→ Gen de susceptibilidad a FNMTC de baja penetrancia en algunas familias |
| HABP2 | 10q25.3 | Variante Gly534GluE en una familia de 7 afectos (45)→ Posible gen de susceptibilidad, no demostrado en otras poblaciones |
| SRRM2 | 16p13.3 | Controvertido (48) |
| Genes del complejo telomere-telomerase complex: TERT etc. | varios | Poca consistencia entre estudios (50,53) |
| RTFC (C14orf93) | 14q11.2 | Variante Val205Met en una familia de 5 afectos (55)→ Posible gen de susceptibilidad, no validado en otros estudios |
| MAP2K5 | 15q23 | Variantes Ala321Thr y Met367Thr presentes en 2 familias (56) → resultados no replicados (57) |
| DUOX2 | 15q21.1 | Variante TyrY1203His en una familia de 6 afectos (58)→ Posible gen de susceptibilidad, no validado en otros estudios |
| NOP53 | 19q13.33 | Variante AspD31His en 3 familias con FNMTC (61)→ Posible gen de susceptibilidad, no validado en otros estudios |
Se han desarrollado diversas estrategias para la búsqueda de genes candidatos responsables del FNMTC. Previamente a la aparición de las técnicas de secuenciación modernas, se habían llevado a cabo:
a) Estudios de ligamiento (linkage). En genética se denomina ligamiento a la asociación física entre dos loci (esto es, su cercanía en una misma hebra de ADN, lo que repercute en una baja frecuencia de recombinación entre ellos durante la meiosis, y, por tanto, a una mayor probabilidad de herencia conjunta). En individuos con parentesco se mapean loci genéticos para encontrar alelos que se hereden ligados a la enfermedad de interés y determinar de manera indirecta la localización cromosómica del gen asociado a FNMTC. Es decir, este tipo de estudios son un método indirecto que usa marcadores genéticos (por ejemplo, microsatélites), ligados al gen de la enfermedad.
Los loci descritos hasta la fecha incluyen TCO28 (19q13.2), fPTC/PRN (1q21), FTEN (8p23.1-p22), NMTC1 (2q21), MNG1 (14q32), 6q22, 8q24 y 4q3229. Sin embargo, los genes candidatos ubicados en estos loci permanecen desconocidos.
b) Estudios de GWA (genome-wide association). Se estudian miles de polimorfismos comparando controles versus pacientes, a fin de encontrar polimorfismos de predisposición. Mediante esta técnica se han identificado, por ejemplo, dos polimorfismos de un solo nucleótido (SNPs), asociados a riesgo de FNMTC: rs944289 (localizado en 14q13.3, cerca de NKX2-1) y rs965513 (localizado en 9q22.33, cerca del gen candidato FOXE1)30. Sin embargo, la mayoría de los estudios están realizados en CDT no familiar31.
c) Estudios dirigidos a genes candidatos o vías metabólicas específicas. Por ejemplo, Pereira et al.32 estudiaron directamente FOXE1 en 60 familias con FNMTC en Portugal, encontrando una asociación entre variantes en línea germinal de este gen y la aparición de FNMTC en estas familias.
Sin embargo, en todos estos estudios los resultados han sido poco fructíferos, describiendo genes que podrían estar implicados en la etiología del FNMTC con baja penetrancia, y únicamente en algunas familias. Además, los resultados no se han replicado en sucesivas investigaciones.
d) Técnicas de secuenciación masiva. En los últimos años, se ha producido un gran avance en el campo de la genómica gracias a la mayor factibilidad, tanto técnica como económica, para realizar la secuenciación masiva del genoma. La secuenciación masiva (next generation sequencing [NGS]) ha conllevado un gran salto ya que ha supuesto pasar de examinar genes específicos a explorar genomas completos. Todo ello, además, de forma mucho más eficiente y rápida que con la secuenciación clásica de Sanger. De este modo se puede comparar, por ejemplo, cómo difiere el genoma de pacientes con una determinada patología frente al genoma de individuos sanos. Desde la incorporación de las nuevas técnicas de secuenciación, se han multiplicado los estudios que tratan de dilucidar las causas genéticas del FNMTC, identificando nuevos genes recogidos en una revisión reciente33. A continuación revisaremos los conocimientos que tenemos acerca de los genes asociados con vulnerabilidad al FNMTC descritos hasta la fecha.
Genes candidatos descritosGenes implicados en la reparación de DNA: ATM (ATM serine/threonine kinase, locus 11q22.3), CHEK2 (checkpoint kinase 2, locus 22q12), XRCC1 (X-Ray repair cross complementing 1, locus 19q13.31) y XRCC3 (X-Ray repair cross complementing 3, locus 14q32).El hecho de que factores como la radiación ionizante aumenten el riesgo de cáncer de tiroides, sugiere que genes reparadores de DNA podrían estar implicados en su fisiopatología. Por ello, se han estudiado genes como ATM, CHEK2, XRCC1 y XRCC3. En varios estudios se ha descrito que determinados polimorfismos en estos genes podrían asociarse con mayor susceptibilidad a CDT, como el rs17879961 en CHEK234, y el rs373759 en ATM35. Además, mutaciones en CHEK2 podrían ser más frecuentes en casos de CDT36. Por el contrario, Ryu et al.37 describieron que el cambio Arg194Trp en XRCC1 confería menor susceptibilidad a CDT en población coreana. Dichas investigaciones se han realizado básicamente comparando pacientes con CDT esporádico versus controles38, no pudiendo establecerse por el momento una clara relación con el FNMTC.
TITF-1 (thyroid transcription factor 1, también conocido como NKX2.1)Ubicado en 14q13, es un factor de transcripción tiroideo. En 2009, se describió la mutación germinal Ala339Val en TITF-1 en cuatro pacientes con historia de BMN y carcinoma papilar de tiroides, dos de los cuales tenían antecedentes familiares de BMN, con o sin cáncer papilar de tiroides39. Un año después, Cantara et al.40 no encontraron en su serie de pacientes con carcinomas papilares de tiroides (CPT), la presencia de esta variante en TITF-1. Por ello no se descarta que esta variante esté asociada con el BMN, pero no puede afirmarse que esté relacionada con el CPT y menos aún con el FNMTC.
FOXE1 (forkhead box E1)Ubicado en 9q22.33, codifica un factor de transcripción tiroideo (TITF-2), que participa en la morfogénesis y migración de la glándula tiroidea. Determinados polimorfismos en este gen se han asociado al FNMTC de manera más robusta. Como hemos comentado, en el estudio de GWA de 2009 de Gudmundsson et al.30 se describieron los polimorfismos rs944289 (localizado en 14q13.3, cerca de NKX2-1) y rs965513 (en 9q22.33, cerca de FOXE1), como predisponentes al cáncer de tiroides en población europea. Bonora et al.41 genotiparon estos y otros 21 SNPs identificados por GWAS en 11 genes candidatos en 672 sujetos pertenecientes a 133 familias con FNMTC de dos o más miembros afectos. Hallaron dos SNPs (rs965513 y rs10759944) en 9q22.33 (cerca de FOXE1), que se asociaban de manera estadísticamente consistente con FNMTC; no así el SNP rs944289. Pereira et al.32 secuenciaron entonces FOXE1 en 60 familias con FNMTC y en 80 casos esporádicos de CPT, encontrando una nueva variante en línea germinal (Ala248Gly) con segregación en una única familia con FNMTC, y que en los estudios funcionales promovía la tumorogénesis.
Por todo, aunque los resultados no son completamente consistentes, FOXE1 podría considerarse un gen de susceptibilidad a FNMTC de baja penetrancia.
SRGAP1 (SLIT-ROBO Rho GTPase-activating protein 1)SRGAP1 (locus 12q14) codifica una proteína que inactiva a CDC42, una pequeña proteína GTPasa de la familia Rho, implicada en la migración neuronal y la tumorogénesis42. Mediante estudios de ligamiento, se identificó el locus 12q14 en 21/38 familias con FNMTC, por lo que posteriormente se procedió a la secuenciación de todos los exones de SRGAP1, ubicados en ese nivel43. Encontraron cuatro mutaciones missense en línea germinal (Gln149His, Ala275Thr, Arg617Cys y His875Arg), cada una en una única familia. Los estudios funcionales demostraron que las variantes Gln149His y Arg617Cys condicionaban pérdida de función de la proteína SRGAP1, que sería incapaz de inactivar a CDC42. Este trabajo sugiere que SRGAP1 podría ser un gen de baja penetrancia asociado a FNMTC. Sin embargo, es necesaria su validación como gen candidato en una cohorte más amplia de familias con FNMTC.
HABP2 (Hyaluronan-Binding Protein 2)HABP2 se localiza en 10q25.3, y codifica una proteasa que se une al ácido hialurónico y participa en la fibrinólisis y la integridad vascular44.
En 2015, aparece el primer estudio que aplica la secuenciación masiva del exoma para el estudio de las bases genéticas del FNMTC. El grupo de Gara45 describió un cambio genético constitucional Gly534Glu en el gen HABP2, que cosegregaba en una familia con siete miembros con CPT, como posible causante de la enfermedad. También describieron este cambio en un 4,7% de 423 casos con CDT esporádico, versus en 0,7% de la población control, lo que indicaba una frecuencia 6,71 veces mayor en pacientes con cáncer de tiroides. Asimismo, demostraron sobreexpresión de la proteína HABP2 en tejido tumoral de los pacientes con FNMTC, en comparación con los de controles y casos esporádicos; además, en los estudios de funcionalidad la presencia de la variante Gly534Glu condicionaba pérdida de función supresora tumoral. Todo ello indicaba que HABP2 podría tener una función supresora tumoral y que el cambio Gly534Glu supondría una predisposición al cáncer de tiroides con penetrancia incompleta. Otro grupo encontró 4/29 familias con FNMTC que también presentaban la misma variante germinal Gly534Glu46. Sin embargo, el papel de HABP2 como gen de susceptibilidad a FNMTC es aún controvertido, dado que estos resultados no se han replicado en otros estudios y la frecuencia poblacional del cambio Gly534Glu podría ser muy variable atendiendo a su origen geográfico47.
SRRM2 (serine/arginine repetitive matrix 2)SRRM2 (locus 16p13.3) codifica una proteína implicada en el splicing. Este gen fue propuesto como candidato en el FNMTC a raíz del estudio de Tomsic en 201548, en el que se sugiere que variantes en esta proteína podrían afectar al splicing de genes implicados en la tumorogénesis tiroidea. Concretamente, llevaron a cabo NGS de dos miembros de una familia de seis afectos de FNMTC, encontrando la variante en heterocigosis c.1037C > T (Ser346Phe,rs149019598), que cosegregaba con el CDT en la familia. Por el contrario, este cambio no fue encontrado cuando analizaron otras 138 familias con CDT y requiere validación ulterior.
Genes del complejo telómero-telomerasaLos telómeros son secuencias repetitivas de ADN no codificante que se encuentran en los extremos de los cromosomas. Su función es dar estabilidad cromosómica, y cuando estas secuencias se acortan, se produce una inestabilidad que favorece la oncogénesis49. Existen varios genes implicados en el mantenimiento de los telómeros, tales como TERT (telomerase reverse transcriptase), o los genes del complejo shelterina (POT1, RAP1, TIN2, TPP1, TRF1 y TRF2)50.
Sabemos que mutaciones somáticas en el promotor de TERT juegan un papel importante en el desarrollo del cáncer de tiroides. Por ello, se ha sugerido que los genes implicados en el mantenimiento de los telómeros podrían estar implicados en CDT, y en el FNMTC en particular. De hecho, se ha descrito que los pacientes con FNMTC tienen los telómeros más cortos que sus familiares no afectos, los controles sanos o casos esporádicos51. Sin embargo, Jendrzejewski et al.52 no encontraron diferencia en la longitud de los telómeros entre CDT esporádicos y FNMTC. Tampoco se han observado diferencias en el número de copias ni en la expresión de diversos genes del complejo telómero-telomerasa en seis familias con FNMTC53, ni se han hallado mutaciones en TERT u otros genes del complejo shelterina en línea germinal en familias con FNMTC50,54. Por tanto, los resultados en la literatura son aún poco consistentes y requieren más investigaciones.
RTFC o C14orf93 (chromosome 14 open reading frame 93)En un estudio de 2017 en China55, combinando análisis de ligamiento y NGS, encontraron la variante Val205Met) en RTFC (locus 14q11.2) en una única familia con cinco miembros con FNMTC. En los estudios funcionales, esta variante promovía la oncogénesis. No se han validado estos resultados en otros estudios independientes.
MAP2K5 (mitogen-activated protein kinase kinase 5)MAP2K5 (también conocido como MEK5), se localiza en 15q23 y codifica una proteína de la familia de las MAP kinasa. En un artículo reciente en población china56, estudiaron a 34 familias de más de ≥ tres miembros afectos de FNMTC mediante NGS, y hallaron dos variantes en el gen MAP2K5 presentes en dos familias: Ala321Thr y Met367Thr). Estas variantes son muy infrecuentes en población sana. Los resultados de los estudios funcionales también sugirieron que MAP2K5 podría ser un gen de susceptibilidad al FNMTC. Sin embargo, otro grupo italiano no replicó estos resultados en 33 familias57, permaneciendo incierta la implicación de MAP2K5 en esta patología.
DUOX2 (dual oxidase 2)Ubicado en 15q21.1, este gen codifica una glicoproteína que participa en el metabolismo del peróxido de hidrógeno (H202), necesario para la actividad de las enzimas tiroideas que participan en la síntesis de hormonas tiroideas. Recientemente, se ha descrito una nueva variante germinal en DUOX2 (la variante missense Tyr1203His), identificada mediante NGS en una familia de seis miembros con FNMTC, y que podría asociarse a susceptibilidad a éste58. La presencia de esta variante aumentaría la fabricación de peróxido de hidrógeno, tóxico para el DNA, promoviendo la tumorogénesis. De manera interesante, la expresión de DUOX2 está aumentada en pacientes homocigotos para el polimorfismo rs965513 en FOXE1, con lo que ambos mecanismos podrían estar conectados, sugiriendo los autores que la desregulación de proteínas implicadas en el metabolismo del H2O2 podría relacionarse con la susceptibilidad al FNMTC.
NOP53 (ribosome biogenesis factor)NOP53 está localizado en 19q13.33 y codifica una proteína nucleolar implicada en la biogénesis ribosómica, regulando la activación de p5359. Está implicado en la vía de señalización PI3K/AKT, una de las más importantes en cáncer de tiroides. De hecho, en un estudio de una familia con tres miembros con FNMTC mediante NGS, se descubrieron tres genes de susceptibilidad (PIK3CB, CAV2 y KANK1), relacionados con la tumorogénesis a través de la vía de PI3K/AKT60.
Con base en un estudio multicéntrico realizado en España de 45 familias con FNMTC, se ha descrito NOP53 como otro gen candidato en el FNMTC. Concretamente, se encontró la variante Asp31His (rs78530808), que cosegregaba en todos los miembros afectos de tres familias con cinco, cuatro y dos miembros afectos de CDT, respectivamente, y no estaba presente en los familiares sanos. Además, los estudios funcionales mostraron una función oncogénica de NOP5361. De hecho, en un estudio previo se había observado mayor expresión de NOP53 en carcinomas foliculares agresivos62.
Por todo ello, NOP53 podría estar implicado en el FNMTC, probablemente como un gen de baja penetrancia, si bien hasta el momento estos recientes hallazgos no se han corroborado en otros estudios.
MicroRNAsTambién se ha hipotetizado que los microRNA, pequeños segmentos de RNA capaces de regular la expresión de otros genes podrían estar implicados en el desarrollo del cáncer de tiroides. En una revisión de 201863, se recogen los distintos microRNA relacionados hasta entonces con los diferentes subtipos de cáncer de tiroides. Destacamos el artículo de Xiong et al.64, por focalizarse en el estudio del perfil de microRNAs del FNMTC, encontrando una desregulación del miR-886-3p y miR-20-a.
LimitacionesTras todo lo expuesto, se pone de manifiesto la complejidad del estudio de la genética del FNMTC por varias razones:
En primer lugar, la dificultad de reclutar datos clínicos y muestras biológicas (sangre y tejido histológico tumoral) de familias que presentan varios casos afectos exige una colaboración multicéntrica. En esta línea, desde la SEEN existe un registro en el que se pueden introducir los FNMTC que encontremos en nuestra práctica clínica (http://www.regcancerdiferenciadofamiliar.es/). Teniendo en cuenta los estudios mencionados, parece que nuestros esfuerzos deberían centrarse en las familias con ≥ tres miembros afectos, para maximizar el componente hereditario. Como hemos comentado, algunas características como la agresividad tumoral y la edad menor al diagnóstico, o la utilidad del cribado, son mucho más claras en las familias de al menos tres personas afectas. Por tanto, parece mejor priorizar la homogeneidad y calidad de las familias reclutadas que la cantidad. Además, dada la alta prevalencia del cáncer de tiroides en población general, no hay que descartar que dentro de una familia con FNMTC pueda haber algún caso de CDT esporádico, es decir, una fenocopia.
En segundo lugar, y vistos los resultados poco consistentes entre los estudios, que sugieren básicamente genes candidatos de baja penetrancia o incluso mutaciones únicas en algunas familias, podría ser que el FNMTC fuese una enfermedad genéticamente heterogénea y poligénica, a diferencia de la mayoría de cánceres hereditarios, donde los genes causantes son habitualmente uno o dos. Esto explicaría por qué no encontramos un nexo común entre las diferentes familias.
Por último, aunque las técnicas de secuenciación masiva del exoma han supuesto un gran salto cualitativo en la bioquímica y genética molecular, y cada vez están más estandarizadas, todavía presentan sus limitaciones y requieren frecuentemente de validación mediante secuenciación de Sanger. Además, los resultados y variantes de interés obtenidas dependen mucho de la estrategia de búsqueda y de los filtros empleados, que son imprescindibles por el volumen ingente de información; esto a la vez implica un sesgo de selección al poder perder por ello variantes potencialmente candidatas que no pueden excluirse completamente65.
ConclusionesHoy en día, el FNMTC continúa siendo una patología heterogénea sin una causalidad genética establecida. No está indicado el estudio genético de las familias con FNMTC al no haber ningún gen claramente establecido como causante del FNMTC, salvo en el caso de los FNMTC asociados a síndromes genéticos hereditarios. Sin embargo, sí es de utilidad la recogida y registro de estas familias para poder llevar a cabo estudios de colaboración multicéntrica y tratar de dilucidar las causas genéticas de su enfermedad.
FinanciaciónEste trabajo no ha recibido ningún tipo de financiación.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
