




El hiperparatiroidismo primario (HPTP) es la principal causa de hipercalcemia. Aunque la enfermedad más común es el HPTP esporádico, por adenoma único, existen formas heredables en un 10% de los casos1. Las variantes hereditarias más frecuentes son parte de síndromes donde el HPTP se asocia a otros trastornos endocrinos, que incluyen la neoplasia endocrina múltiple (MEN) tipo 1 y 2, el síndrome hiperparatiroidismo asociado a tumor de mandíbula (HPT-JT), la hipercalcemia hipocalciuria familiar (HHF) y el hiperparatiroidismo familiar aislado (HPFA).
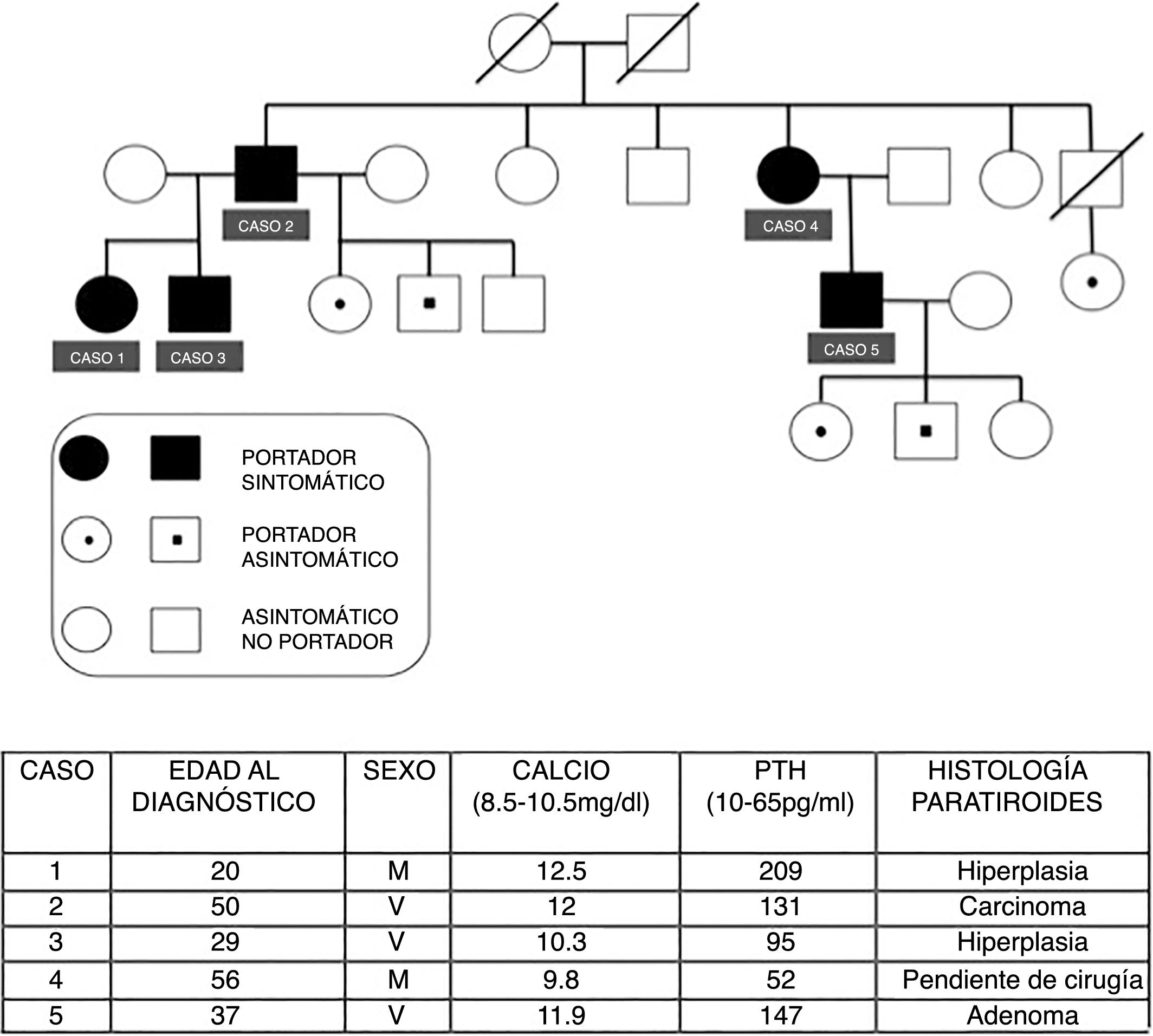
Presentamos los casos de una familia con HPFA, cuyo caso índice (caso 1) fue valorado por primera vez en 2006. Desde entonces se han estudiado un total de 3 generaciones, 15 miembros de la familia, 10 son portadores de la mutación en el gen HRPT2 y 5 de ellos son portadores sintomáticos. En la figura 1 se describe el árbol genealógico de la familia y se resumen las principales características clínicas, analíticas e histológicas de los pacientes afectos.

Caso 1: mujer de 20 años que consultó por HPTP, con nefrolitiasis y osteopenia. La gammagrafía MIBI fue negativa en 2 ocasiones. Se realizó paratiroidectomía inferior derecha, informando intraoperatoriamente de hiperplasia paratiroidea. La paratohormona (PTH) intraoperatoria descendió adecuadamente según los criterios de Miami2. Tras la cirugía, mantiene niveles normales de PTH y calcio.
Caso 2: varón de 50 años, con síntomas de nefrolitiasis. La analítica objetivó hipercalcemia, hipofosfatemia y elevación inapropiada de PTH, con gammagrafía MIBI negativa. Sin evidencia de enfermedades endocrinas asociadas. En la cirugía se halló paratiroides superior izquierda aumentada de tamaño y resto de glándulas sin alteraciones. Se practicó paratiroidectomía superior izquierda e intraoperatoriamente se comprobó el descenso de PTH. La histología definitiva informó de carcinoma paratiroideo. Con adecuados controles posteriores.
Al encontrar 2 miembros con HPTP en la misma familia, se realizó estudio molecular que incluyó análisis del gen HRPT2 (1q25-q31) en muestra de sangre periférica mediante secuenciación automática, utilizando cebadores específicos de ADN que flanquean la región donde se localiza la mutación responsable de la enfermedad en esta familia. Este estudio mostró una mutación en heterocigosis en el exón 6, c.456_459dup/p.Ala154IlefsX16, en los 2 primeros casos y en el resto de los portadores.
Caso 3: varón de 29 años, con hiperparatiroidismo normocalcémico. La gammagrafía MIBI mostró un adenoma paratiroideo ectópico. Se realizó paratiroidectomía subtotal más timectomía. La anatomía patológica informó de hiperplasia paratiroidea.
También se realizó estudio genético a los hermanos del padre, siendo negativo para 5 de ellos y positivo en un caso (caso 4).
Caso 4: mujer de 56 años. Actualmente en estudio por osteopenia, cólicos nefríticos y déficit de vitamina D, pendiente de completar estudio y posterior intervención si precisara.
Caso 5: varón de 37 años, con HPTP sintomático asociado a microlitiasis y osteopenia en fémur. Recientemente se practicó paratiroidectomía subtotal con timectomía. La histología informó de adenoma paratiroideo inferior derecho y resto de glándulas hiperplásicas. Tiene 3 hijos, 2 de ellos portadores del gen.
Los portadores asintomáticos de la mutación familiar, siguen control anual con analítica, ecografía cervical y renal.
El HPFA se considera una entidad autónoma no sindrómica o una expresión incompleta de uno de los síndromes genéticos causantes de HPTP. El diagnóstico es de exclusión y requiere la presencia de al menos 2 parientes de primer grado con HPTP y ausencia de otras manifestaciones endocrinas1, como la enfermedad hipofisaria y pancreática características del MEN-1 o los tumores fibrosos de mandíbula y riñón del HPT-JT3. Si el HPFA es una variante o una etapa precoz del síndrome MEN-1, aún no se ha establecido.
Normalmente aparecen en edades más tempranas, entre los 20-25 años, unos 30 años antes que en el HPTP esporádico, como nuestro caso índice. Sufren afectación multiglandular, menor tasa de curación y mayor riesgo de HPTP recurrente y carcinomas, en comparación con el HPTP esporádico1,4. Además, con mayor frecuencia, desarrollan hipercalcemia grave, en comparación con los pacientes afectos de MEN-13.
Se hereda de forma autosómica dominante, sin existir un gen específico. Sin embargo, se han descrito mutaciones germinales en MEN-1, HRPT2 y CASR en un número significativo de familias, por lo que mutaciones del mismo gen pueden ser responsables de diferentes síndromes1,5.
La forma clínica de presentación es más grave y la histología suele ser compatible con adenomas o carcinomas, como en nuestro caso, siendo menos frecuente la aparición de hiperplasias. Sin embargo, ningún paciente mostró cambios quísticos en la anatomía patológica, típico del HPT-JT1.
Recientemente, se ha descubierto una mutación germinal en GCM-2 en familias con HPFA con niveles más elevados de paratohormona, más riesgo de enfermedad multiglandular, de carcinoma y, menor tasa de curación bioquímica6,7.
Por otro lado, en el 15-20% de los carcinomas paratiroideos esporádicos se identifica la mutación del gen HRTP2 (CDC73), por lo que algunos autores4,5 lo consideran un gen supresor de tumores. Los pacientes con carcinoma paratiroideo de reciente diagnóstico deben someterse a una cuidadosa revisión de la historia familiar y, se les debería ofrecer estudio genético de la mutación del gen HRPT23.
Los portadores asintomáticos precisan una vigilancia prospectiva óptima, con ecografía cervical y determinación periódica de calcio sérico y PTH, para conseguir detectar precozmente la enfermedad8.
Mientras que el manejo quirúrgico del HPTP esporádico está bien establecido, el tratamiento de elección en pacientes con HPFA asociados con HRPT2 sigue siendo controvertido. Varios autores8,9 argumentan que, dado el alto índice de recurrencia o persistencia de la enfermedad, la morbilidad que supone una segunda reintervención y el riesgo de desarrollar carcinoma paratiroideo8,10, la paratiroidectomía subtotal debería convertirse en el enfoque inicial para los pacientes con la mutación del gen HRPT2. Otros autores sugieren que la paratiroidectomía total es el tratamiento óptimo, aún en ausencia de sospecha de cáncer3. Nosotros somos partidarios de realizar una paratiroidectomía subtotal con timectomía, para evitar el desarrollo de carcinomas paratiroideos.
En los casos de HPTP de inicio en adultos jóvenes, sobre todo si asocian antecedentes familiares de HPTP y ausencia de otros cuadros sindrómicos es importante realizar estudios genéticos ampliados a otras mutaciones, además de MEN-1, incluyendo HRPT2. En las familias con HPFA y mutación del gen HRPT2 es crucial el seguimiento estrecho de los portadores asintomáticos y se recomienda un abordaje quirúrgico más agresivo en los casos con HPTP por el mayor riesgo de desarrollo de carcinoma de paratiroides.
A la Dra. M. Robledo del Centro Nacional de Investigaciones Oncológicas por el estudio genético.
