




La hipofosfatemia ligada al cromosoma X (XLH), aun entrando en la categoría de enfermedades raras, es la forma más común del grupo heterogéneo de trastornos hereditarios que se conocen como raquitismo hipofosfatémico. Su prevalencia es de 1:20.0001, y tiene su origen en la mutación del gen PHEX, localizado en el brazo corto del cromosoma 22, que codifica para una endopeptidasa reguladora de fosfato de nombre homólogo. Se han descrito cientos de variedades de mutaciones2 que codifican para proteínas PHEX anómalas que pierden su función de inhibición sobre el factor de crecimiento fibroblástico 23, lo que suprime la expresión del cotransportador tubular sodio-fósforo, y con ello, el descenso de la reabsorción tubular de fosfato e hipofosfatemia3. Además, el factor de crecimiento fibroblástico 23 suprime los niveles y actividad de la α-1-hidroxilasa, lo que lleva a una disminución en los niveles de la 1,25-dihidroxivitamina D, forma activa de la vitamina D.
La enfermedad tiene una amplia variabilidad fenotípica y sus signos y síntomas incluyen: raquitismo, osteomalacia, talla baja, arqueamiento de extremidades inferiores, craneosinostosis, dolores óseos, fracturas, calcificaciones extraóseas, defectos en la dentina y sordera neurosensorial, entre otros. Los estudios han demostrado una importante afectación en la calidad de vida de estos pacientes con reducción en la movilidad e incapacidad funcional4,5.
El tratamiento clásico de la XLH se ha basado en el suplemento oral de fósforo y calcitriol, tratamiento con poca adherencia de los pacientes por su mala tolerancia digestiva y que no está exento de importantes efectos adversos como nefrocalcinosis o hiperparatiroidismo secundario6. Sin embargo, un nuevo anticuerpo monoclonal humano antifactor de crecimiento fibroblástico 23, burosumab, aprobado por la EMA y la FDA, y ya en uso compasivo pediátrico en nuestro país, ha abierto la posibilidad de bloquear el mecanismo de la enfermedad y asegurar un desarrollo normal en la infancia7 y cuyo papel en el adulto está siendo estudiado8.
En esta carta presentamos 2 casos con mutaciones no descritas hasta la fecha.
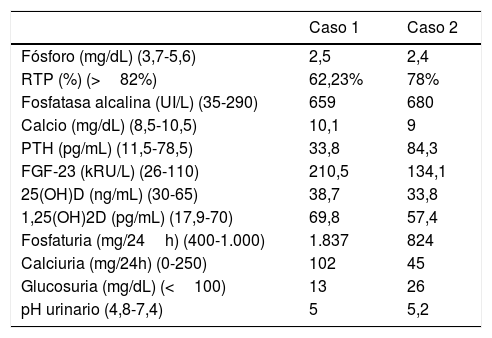
Caso clínico 1Niño de 2 años y 8 meses remitido por genu varo bilateral desde los 9 meses de edad, hasta entonces en seguimiento exclusivamente por traumatología, sin otros antecedentes médicos destacables. Se realizó una analítica sanguínea y de orina (tabla 1) con hallazgos compatibles con XLH y una ecografía renal que resultó anodina, por lo que se solicitó estudio genético e inició tratamiento con fósforo oral y calcitriol.
Hallazgos de laboratorio al diagnóstico
| Caso 1 | Caso 2 | |
|---|---|---|
| Fósforo (mg/dL) (3,7-5,6) | 2,5 | 2,4 |
| RTP (%) (>82%) | 62,23% | 78% |
| Fosfatasa alcalina (UI/L) (35-290) | 659 | 680 |
| Calcio (mg/dL) (8,5-10,5) | 10,1 | 9 |
| PTH (pg/mL) (11,5-78,5) | 33,8 | 84,3 |
| FGF-23 (kRU/L) (26-110) | 210,5 | 134,1 |
| 25(OH)D (ng/mL) (30-65) | 38,7 | 33,8 |
| 1,25(OH)2D (pg/mL) (17,9-70) | 69,8 | 57,4 |
| Fosfaturia (mg/24h) (400-1.000) | 1.837 | 824 |
| Calciuria (mg/24h) (0-250) | 102 | 45 |
| Glucosuria (mg/dL) (<100) | 13 | 26 |
| pH urinario (4,8-7,4) | 5 | 5,2 |
FGF-23: factor de crecimiento fibroblástico 23; PTH: parathormona; RTP: reabsorción tubular de fosfato; 25(OH)D: 25-hidroxivitamina D; 1,25(OH)2D: 1,25-dihidroxivitamina D.
El estudio de secuenciación del gen PHEX identificó la mutación c.2182C>T; p.Gln728Ter, un codón de parada en aparente heterocigosis, pese a encontrarse en el cromosoma X de un varón. Se confirmó que el paciente tenía un único cromosoma X (por cariotipo [46 XY]) y un único alelo del gen PHEX (por método de amplificación múltiplex con sonda dependiente de ligado [MLPA]). El estudio genético realizado a la madre no detectó la variante estudiada en el gen PHEX, por lo que se clasificó como mutación de novo. Además, hasta donde alcanza nuestro conocimiento, esta es una variante no descrita todavía en la literatura. La aparente heterocigosis se encontró en ADN procedente de sangre periférica y de saliva, por lo que se encuentra en mesodermo y ectodermo y se observa tanto por secuenciación de última generación (NGS) como por método de Sanger.
Durante el seguimiento, al paciente se le tuvo que realizar osteotomía y colocación de placas para corregir las importantes deformidades en miembros inferiores.
Caso clínico 2Mujer de 18 años, diagnosticada de XLH a los 2 años y 7 meses (tabla 1). Ya entonces destacaba que su madre, también afecta, presentaba una talla baja extrema y marcado arqueamiento de miembros inferiores, mientras que ella tenía poca morbilidad, con un posterior desarrollo estaturoponderal y esquelético normales.
Se le realizó estudio genético por MLPA identificando un cariotipo con una trisomía 47XXX en mosaico mayoritario (22/25 metafases) respecto a 46XX, identificando 3 copias del gen PHEX, excepto en el exón 16 de este, del que se detectan solo 2. Este resultado, compatible con una deleción de dicho exón en una de las 3 copias, también se encontró en el análisis genético de la madre, por lo que probablemente sea la causa de la enfermedad. El hecho de que la paciente presente una trisomía X y que la deleción solo se encuentre en una de las 3 copias podría explicar la escasa expresividad clínica.
Desde el diagnóstico sigue tratamiento con calcitriol y fósforo oral, reconociendo un seguimiento errático en la adolescencia, pero actualmente adecuado. En estos años ha desarrollado un hiperparatiroidismo secundario y una incipiente nefrocalcinosis.
En conclusión, la XLH es una entidad con una amplia variabilidad en su expresión clínica, sin relación clara genotipo-fenotipo que requiere de un equipo multidisciplinar de especialistas para su manejo. No podemos olvidar que hay formas paucisintomáticas, de difícil diagnóstico en ausencia de familiares afectos, sobre todo si se manifiestan en la edad adulta con dolor óseo crónico, fracturas o seudofracturas, osteoartritis precoz, problemas dentales o entesopatías.
FinanciaciónLa presente investigación no ha recibido financiación alguna.
Conflicto de interesesLos autores declaran que no existe ningún potencial conflicto de interés relacionado con este artículo.
