





Monogenic diabetes caused by changes in the gene that encodes insulin (INS) is a very rare form of monogenic diabetes (<1%). The aim of this work is to describe the clinical and glycaemic control characteristics over time from four members of a family diagnosed with monogenic diabetes with the novel mutation: c.206del,p.(Gly69Aalfs*62) located in exon 3 of the gene INS. 75% are females, with debut in adolescence and negative autoimmunity. In all cases, C-peptide is detectable decades after diagnosis (>0.6ng/ml). Currently, patients are being treated either with insulin in a bolus-basal regimen, oral antidiabetics or hybrid closed loop system. Monogenic diabetes due to mutation in the INS is an entity with heterogeneous presentation, whose diagnosis requires high suspicion and presents an important clinical impact. Given the lack of standards in this regard, therapy must be individualized, although insulin therapy could help preserve beta cell functionality in these subjects.
La diabetes causada por cambios en el gen que codifica la insulina (INS) es una forma rara de diabetes monogénica (<1%). El objetivo del presente trabajo fue describir las características clínicas y de control glucémico a lo largo de la evolución de 4 miembros de una familia diagnosticados de diabetes monogénica con la mutación no descrita: c.206del,p.(Gly69Aalfs*62) localizada en el exón 3 del gen INS. El 75% fueron mujeres, con inicio en la adolescencia y autoinmunidad negativa. En todos los casos, el péptido C fue detectable décadas después del diagnóstico (>0,6ng/ml). Actualmente, los pacientes reciben tratamiento con insulina en un régimen de bolo basal, antidiabéticos orales o un sistema híbrido de circuito cerrado. La diabetes monogénica por mutación en el INS es una entidad de presentación heterogénea, cuyo diagnóstico requiere alta sospecha y presenta un impacto clínico importante. Ante la falta de estándares al respecto, la terapia debe ser individualizada, aunque el tratamiento con insulina podría ayudar a preservar la funcionalidad de las células beta en estos sujetos.
Although insulin was discovered more than a century ago and its role in the etiopathogenesis of diabetes mellitus is well known.1 However, it was not until 2008 that mutations in the gene encoding insulin were first identified as the cause of permanent neonatal diabetes.2 In the last decade, the number and type of mutations identified have been increasing, allowing the study of new diagnostic and therapeutic perspectives for this type of monogenic diabetes.3
The aim of the present study was to describe the clinical expression of a family diagnosed with diabetes due to a dominant mutation in the insulin gene, as well as its evolution over time and its response to different treatments during follow-up in the Paediatrics and Endocrinology and Nutrition Departments of a tertiary hospital.
Description of the caseThis is a family with 4 subjects affected by monogenic diabetes due to a mutation in the insulin gene (INS) formed by: two siblings: a 74-year-old woman (subject 1) and a 69-year-old man (subject 2), the latter being the father of a 33-year-old woman (subject 3) and another 31-year-old woman (subject 4). Fig. 1 shows the family tree of the family studied.
 – in heterozygosis across three generations.")
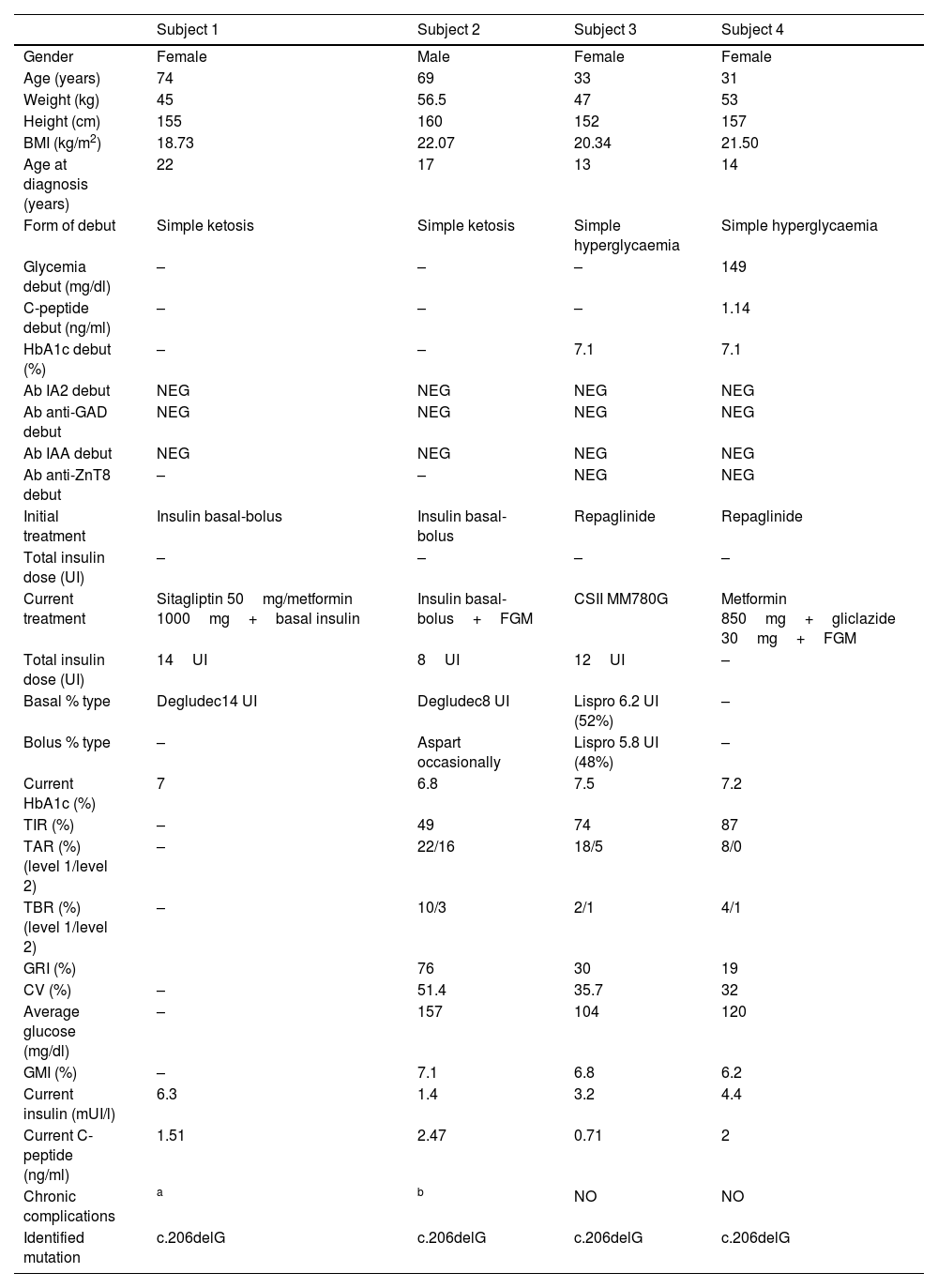
Table 1 summarizes the clinical, anthropometric, analytical, glucometric and genetic characteristics of the 4 patients in our study. All patients in the study debuted with diabetes during adolescence/early adulthood, 50% with simple ketosis and 50% with simple hyperglycaemia. Autoimmunity (anti-glutamate decarboxylase (anti-GAD)), anti-tyrosine phosphatase (IA-2), anti-insulin (IAA) and anti-zinc transporter 8 antibodies (anti-ZnT8) was negative in all study cases. Fifty percent started treatment with sulfonylureas, while the other 50% started treatment with insulin therapy (bolus-basal regimen). The current BMI (Body Mass Index) in the study patients was between 18.73 and 22.07kg/m2.
Clinical, glycometric and genetic characteristics of the studied family.
| Subject 1 | Subject 2 | Subject 3 | Subject 4 | |
|---|---|---|---|---|
| Gender | Female | Male | Female | Female |
| Age (years) | 74 | 69 | 33 | 31 |
| Weight (kg) | 45 | 56.5 | 47 | 53 |
| Height (cm) | 155 | 160 | 152 | 157 |
| BMI (kg/m2) | 18.73 | 22.07 | 20.34 | 21.50 |
| Age at diagnosis (years) | 22 | 17 | 13 | 14 |
| Form of debut | Simple ketosis | Simple ketosis | Simple hyperglycaemia | Simple hyperglycaemia |
| Glycemia debut (mg/dl) | – | – | – | 149 |
| C-peptide debut (ng/ml) | – | – | – | 1.14 |
| HbA1c debut (%) | – | – | 7.1 | 7.1 |
| Ab IA2 debut | NEG | NEG | NEG | NEG |
| Ab anti-GAD debut | NEG | NEG | NEG | NEG |
| Ab IAA debut | NEG | NEG | NEG | NEG |
| Ab anti-ZnT8 debut | – | – | NEG | NEG |
| Initial treatment | Insulin basal-bolus | Insulin basal-bolus | Repaglinide | Repaglinide |
| Total insulin dose (UI) | – | – | – | – |
| Current treatment | Sitagliptin 50mg/metformin 1000mg+basal insulin | Insulin basal-bolus+FGM | CSII MM780G | Metformin 850mg+gliclazide 30mg+FGM |
| Total insulin dose (UI) | 14UI | 8UI | 12UI | – |
| Basal % type | Degludec14 UI | Degludec8 UI | Lispro 6.2 UI (52%) | – |
| Bolus % type | – | Aspart occasionally | Lispro 5.8 UI (48%) | – |
| Current HbA1c (%) | 7 | 6.8 | 7.5 | 7.2 |
| TIR (%) | – | 49 | 74 | 87 |
| TAR (%) (level 1/level 2) | – | 22/16 | 18/5 | 8/0 |
| TBR (%) (level 1/level 2) | – | 10/3 | 2/1 | 4/1 |
| GRI (%) | 76 | 30 | 19 | |
| CV (%) | – | 51.4 | 35.7 | 32 |
| Average glucose (mg/dl) | – | 157 | 104 | 120 |
| GMI (%) | – | 7.1 | 6.8 | 6.2 |
| Current insulin (mUI/l) | 6.3 | 1.4 | 3.2 | 4.4 |
| Current C-peptide (ng/ml) | 1.51 | 2.47 | 0.71 | 2 |
| Chronic complications | a | b | NO | NO |
| Identified mutation | c.206delG | c.206delG | c.206delG | c.206delG |
BMI: body mass index; HbA1c: glycated haemoglobin A1c; Ac: antibodies; IA2: anti-tyrosine phosphatase; anti-GAD: anti-glutamate decarboxylase; IAA: anti-insulin; anti-ZnT8: anti-zinc transporter 8; TIR: time in range; TAR: time above range; TBR: time below range; CV: coefficient of variation; GMI: glucose management indicator, GRI: glycemia risk index; FGM: flash glucose monitoring; CSII: continuous subcutaneous insulin infusion; NEG: negative.
Negative values of anti-GAD antibodies, IA2, IAA and anti-ZnT8 below 1.00U/ml were considered.
The index case from which the diagnosis of monogenic diabetes subtype INS was made was subject 3. Clinical suspicion was established based on the absence of overt ketoacidosis, the negativity of autoimmunity and the presence of a family history (although poorly labelled at the time) of diabetes mellitus. All 4 subjects in the same family had the same heterozygous mutation in the coding region of the INS gene (exon 3), consisting of a deletion of a guanine (c.206del).
This mutation results in a change in the reading pattern from amino acid 69 of the C-peptide region: p.(Gly69Alafs*62) and a longer protein. According to the American College of Medical Genetics and Genomics (ACMG) guidelines for variant classification, this alteration is considered pathogenic. It is a frameshift null variant, i.e. with a change in the reading pattern (PVS1_Very Strong), with a null allele frequency since it has not been reported in the gnomAD population database (PM2_Moderate), and the variant is consistent with the patient's phenotype and family history, since it co-segregates in the family in cases with diabetes (PP4_Supporting). This variant has not been previously described in the scientific literature.
Case managementThey are currently 75% on insulin treatment, while subject 4 receives only oral antidiabetics, in this case Metformin 850mg and Gliclazide 30mg. The latter uses intermittently scanned Continuous Glucose Monitoring (isCGM) (Free Style Libre, Abbott Diabetes Care, Whitney, UK). Of those on insulin therapy, one patient (subject 1) is treated with basal insulin along with oral antidiabetics (Metformin 1000mg and Sitagliptin 50mg, without isCGM), one patient (subject 2) uses bolus-basal regimen along with isCGM, and the remaining patient (subject 3) uses a hybrid closed-loop system (MiniMed™ 780G System, Medtronic, Northridge, California, USA). The patients’ current HbA1c is between 6.8 and 7.5% and current C-peptide between 0.71 and 2.47ng/ml. The isCGM parameters differed between subjects, from the best glycaemic control defined by the lower GRI (Glycemia Risk Index)4 in the patient treated with oral antidiabetics (subject 4), to the worst control in the oldest patient in the series (subject 2) on basal-bolus insulin therapy. Both the latter and his sister (subject 1) present chronic micro and macrovascular complications at this moment (50% of the series).
Clinical guidelinesMonogenic diabetes mellitus due to mutations in the insulin gene (former called MODY 10), despite its low prevalence,5 has a great clinical importance, both for its contribution to the knowledge of the physiology of insulin secretion and for the possible therapeutic repercussions of its diagnosis and great clinical variability.
The INS is located on chromosome 11p15.5. It consists of 3 exons separated by two introns, of which only exons two and three are protein coding.3 Insulin biosynthesis begins with the translation of the messenger RNA into preproinsulin in pancreatic beta cells. The molecule is then transported from the cytosol to the endoplasmic reticulum, acquiring its three-dimensional structure through folding and the formation of three disulfide bridges. This region is of special importance since it is where most diabetes-causing mutations have been identified. Subsequently, the correctly folded proinsulin passes to the Golgi apparatus, where it is stored in secretory granules for its maturation and subsequent excretion by β-cells in response to different metabolic signals.6
Mutations in the INS can be dominant (in heterozygosis) or recessive (in homozygosis or compound heterozygosis). Dominant mutations are the most common, occur in the coding region and generally result in an incorrectly folded proinsulin molecule that becomes trapped and accumulates in subcellular compartments and eventually leads to oxidative stress/apoptosis destruction of pancreatic β-cells with consequent insulinopenia. On the other hand, recessive mutations generally occur in the regulatory regions and usually originate a non-functioning proinsulin by loss of function or reduction of its synthesis, without producing β-cell destruction.3
Although clinically has been described as a typical neonatal onset diabetes (generally in the first 4–6 months of life), it is not uncommon to be diagnosed during childhood and even in adulthood. Diagnosis requires suspicion and identification of the pathogenic mutation responsible by genetic analysis by sequencing of the insulin gene. Although suspicion could be straightforward in those with neonatal diabetes7; in those with late-onset forms, its manifestations do not differ from those of type 1 diabetes (T1D), so the diagnostic will be established in patients with high familial burden and absence of diabetes-associated autoantibodies and HLA risk among others.7,8 In fact, different clinical expression (including the age of onset) has been described for the same INS mutation.5,9
Areas of uncertaintyThe present study shows clinical variability in four subjects from two generations of a family with a very rare form of monogenic diabetes. The diagnosis of monogenic diabetes due to mutations in the INS usually occurs in the neonatal period (neonatal monogenic diabetes)5,9; however, in all the cases in our series, diabetes was diagnosed during late childhood and early adulthood. INS-MODY type diabetes is a rare form within MODY (<1%), with HNF1A-MODY (30–50%), GCK-MODY (30–50%), HNF4A-MODY (10%), and HNF1B-MODY (1–5%) being more frequent.10,11
In our series the age of diagnosis overlaps with the period of onset of T1D, there being no case of diagnosis in the neonatal period unlike those reported in other series.11 Given the diagnostic complexity, a high degree of clinical suspicion is required, based mostly on the presence of high familial aggregation accompanied by the absence of autoimmunity (anti-GAD, IA-2, IAA and anti-ZnT8 antibodies).12–14 However, not all patients with T1D possess these pathophysiological features nor is the diagnosis obvious. Although classification as T1Da (autoimmune) corresponds to 70–90% of cases, there are around 10% of idiopathic forms (T1Db) whose specific pathogenesis is still unclear,15 which makes it difficult to differentiate from other types of monogenic diabetes, especially the less frequent ones such as INS mutations. On the other hand, 1% of patients with monogenic diabetes may present positive antibodies,16 although in our series autoimmunity was negative in all cases.
The high familial aggregation should be another feature that leads us to suspect monogenic diabetes. Generally, a family history of diabetes is observed in two or three consecutive generations with affected subjects in different generations of the family, as it is the case in our series. However, it should not be forgotten that in the case of T1D about 10–15% of patients have a family member with the disease, and this is even higher in type 2 diabetes (T2D).17,18
Other determinations such as C-peptide have also a role in the differential diagnosis. In fact, values below 0.6ng/ml more than 3 years after diagnosis are highly suggestive of T1D.19 However, in monogenic diabetes due to alterations in the INS gene, great clinical and analytical variability is described between different INS mutations at diagnosis and even between patients affected by the same mutation.20,21 This heterogeneity in the form of debut and analytical parameters at diagnosis (glycemia, HbA1c, glucometric parameters) in our series is consistent with what has been previously described. Furthermore, all the patients in our series had detectable C-peptide levels (>0.6ng/ml).
Regarding follow-up, two of the four subjects present chronic complications. These are two sibling patients and the oldest in our series, with microvascular and macrovascular complications including diabetic retinopathy, diabetic nephropathy, chronic ischaemic heart disease, peripheral artery disease and diabetic foot. This may be due to the significant heterogeneity between different mutations and even in patients with the same mutation in terms of long-term clinical evolution and the possibility of developing microvascular and macrovascular complications.
There are no specific recommendations on treatment in monogenic diabetes due to INS mutation, but it is postulated that the use of insulin therapy could decrease the endogenous demand for this hormone, reducing the toxic effect of altered insulin on β-cells, thus helping to preserve their function. This, in turn, could help preserve a pool of viable β cells that would be the basis for future treatments with agents that support insulin expression and biosynthesis from the healthy allele of the INS.3 However, non-insulin therapeutic regimens with sulfonylureas could have utility in achieving acceptable glycaemic control, although there is insufficient evidence of their long-term effect or of other therapies with theoretical advantages over β-cell preservation.3,7,8 In a review carried out by Aarthy et al.,22 27 cases of patients with mutation in the insulin gene were included, who receive different treatments: diet, metformin, acarbose, sulfonylureas and insulin therapy with a single case of ISCI. As in our series, treatment heterogeneity is shown, from patients treated only with oral drugs to one patient with a hybrid closed loop system and another receiving treatment with a dipeptidyl peptidase 4 inhibitor (sitagliptin), with no other precedents found in the literature to our knowledge so far.
In the patients in our study, the presence of diabetes in all generations suggested an autosomal dominant inheritance, confirmed genetically later and with the finding of a genetic alteration not previously described in the literature, located in exon 3 (coding for the protein). The index case belongs to the youngest generation, subsequently giving rise to the detection of other affected relatives with the same mutation, some previously erroneously labelled as T1D (subjects 1 and 2), which indicates the importance of screening in relatives of patients diagnosed with monogenic diabetes.11 For the descendants of affected cases, genetic screening for the disease in childhood should be considered taking into account the heterogeneity in the described cases.
Although the present study and its cross-sectional design may suffer from a limited number of cases, it evaluates a pathology with a very low prevalence, with different cases belonging to several generations of a family and with exhaustive knowledge of the subjects and their variables (included genetic, diabetes complications and CGM.3,11,22
Conclusion and recommendationsMonogenic diabetes due to mutation in the INS is an entity with low prevalence and heterogeneous presentation, whose clinical suspicion should be based on the absence of autoimmunity in young subjects with a high family burden. The diagnosis of this entity has an important clinical repercussion, both for its contribution to the knowledge of the physiology of insulin secretion and for the possible therapeutic consequences of its diagnosis. Given the absence of standards in this regard, therapy should be individualized, although the use of insulin therapy could help to preserve beta cell functionality in these subjects.
Conflict of interestThe authors declare no conflict of interest.
