




A 45-year-old woman was referred by her general practitioner to the Endocrinology Division due to hypertriglyceridaemia and hair loss. The patient presented a Cushingoid phenotype, which began in adolescence, with a marked absence of adipose tissue in the limbs and gluteofemoral region and an accumulation of fat in the face, neck, axillae, supraclavicular region and upper back. She had a Herculean appearance despite leading a sedentary life, with marked musculature in the upper and lower limbs and calf hypertrophy (Fig. 1A–C). She also presented mild acanthosis nigricans in the neck and axillae. At that time, the patient was not diagnosed with diabetes, although she did have high blood pressure and a lipoma on her back had been removed. Otherwise, she did not present symptoms suggestive of cardiac, muscular or neurological involvement, nor did she have a history of acute pancreatitis. Since menarche, her menstrual cycles had always been irregular. However, she did not have hirsutism. Apart from the above, physical examination did not reveal anything remarkable. The body composition analysis, evaluated by DXA, showed: total fat 22.1%; upper-limb fat 15.4%; lower-limb fat 11.4%; and trunk fat 29.5%.
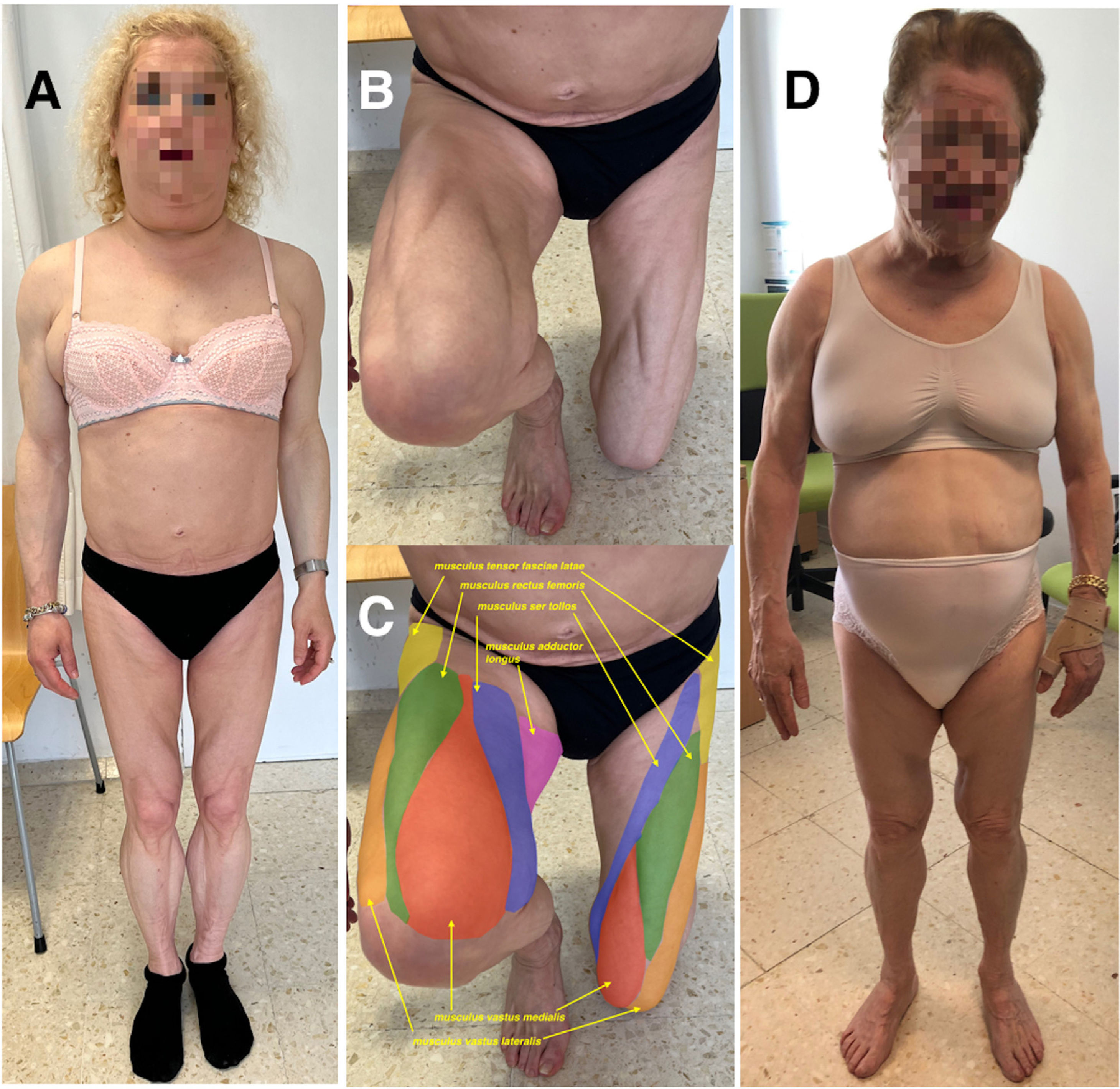
 Index case. A 50-year-old woman with a singular phenotype characterised by an absence of subcutaneous adipose tissue in the hips and limbs, an accumulation of fat in the face, neck and axillae and marked muscle hypertrophy. (B) Lower limbs of the index case showing the striking musculature of the thighs. (C) The same photo as panel B in which the different muscles of the thighs have been coloured and identified (arrows). (D) Mother of the index case, 72 years old, with a similar phenotype. Both patients are carriers of the variant c.1396A>G (p.Asn466Asp) in the LMNA gene, which is diagnostic of type 2 familial partial lipodystrophy.")
(A) Index case. A 50-year-old woman with a singular phenotype characterised by an absence of subcutaneous adipose tissue in the hips and limbs, an accumulation of fat in the face, neck and axillae and marked muscle hypertrophy. (B) Lower limbs of the index case showing the striking musculature of the thighs. (C) The same photo as panel B in which the different muscles of the thighs have been coloured and identified (arrows). (D) Mother of the index case, 72 years old, with a similar phenotype. Both patients are carriers of the variant c.1396A>G (p.Asn466Asp) in the LMNA gene, which is diagnostic of type 2 familial partial lipodystrophy.
At the age of 50, her blood tests revealed prediabetes (A1c 6.0%) with a basal blood glucose of 123mg/dl, triglycerides of 180mg/dl, ALT: 119IU/l [5–40], AST: 70IU/l [5–40], GGT: 393IU/l [5–36], plasma leptin: <1ng/ml [20–37], insulinaemia: 28.1μIU/ml [4.7–14.6], and HOMA-IR: 5.4 [1–3.3]. Abdominal ultrasound ruled out hepatic steatosis. Since laminopathies can be associated with cardiac abnormalities,1 an echocardiogram and Holter monitoring were performed, which did not show structural heart disease or heart rhythm disturbances.
Her 72-year-old mother, diagnosed with type 2 diabetes mellitus at 45, with poor metabolic control (A1c 11.9%), had a similar phenotype, although muscle hypertrophy was not so evident (Fig. 1D). She did not have acanthosis nigricans and at the age of 76 she underwent an infracondylar amputation due to peripheral arterial disease. At present, she is on insulin, metformin, atorvastatin and aspirin.
Given the patient's singular phenotype, highly suggestive of familial partial lipodystrophy, the LMNA gene (NM_170707.3) was sequenced, finding the variant c.1396A>G (p.Asn466Asp) in heterozygosis. This variant was also present in her mother. They were therefore diagnosed with type 2 familial partial lipodystrophy or Dunnigan disease (MIM #151660).2
These cases are a paradigmatic example of a rare disease that had gone unnoticed over the years, despite the fact that the phenotype had always been very striking. The marked muscle hypertrophy in the index case, making it possible to accurately distinguish most of the thigh muscles (Fig. 1B, C), is associated with a conspicuous absence of adipose tissue in certain parts of the body and an abnormal accumulation of fat in others, as well as with signs of insulin resistance. Such characteristics should alert the clinician to this type of rare disease, which follows an autosomal dominant inheritance pattern, leading to an investigation of the family history.
Familial partial lipodystrophy was first described in 1975,3 although the causes were not identified until 2000, when the first gene related to these disorders (LMNA) was found.4 Currently, six genes responsible for these subtypes of lipodystrophies are known5 (LMNA, PPARG, PLIN1, LIPE, CIDEC, AKT2), with dominant or recessive inheritance patterns, although most cases correspond to subtype 2. These patients usually have metabolic (non-ketogenic diabetes mellitus, hypertriglyceridaemia) and liver comorbidities, which have been related to an ectopic deposition of fat in the muscles and liver.6 Treatment of this condition is that of its associated comorbidities. Diet and physical exercise are the cornerstone of its management, albeit taking particular care regarding exercise due to the risk of associated rhythm disturbances or structural cardiomyopathy.2 As for the pharmacological treatment of diabetes, the current multi-society guidelines recommend the use of metformin and, if necessary, insulin. Other antidiabetic drugs could be used. However, there are no large studies endorsing their efficacy. For dyslipidaemia, the same guidelines recommend statins as the first choice of treatment. These can be associated with fibrates and/or omega-3 fatty acids in cases of severe hypertriglyceridaemia.7 Recombinant human leptin, associated with diet, could be a therapeutic option in selected cases if conventional treatment fails.8
Beyond the effect that the absence of subcutaneous fat has on muscle definition, the mechanisms responsible for the marked muscle hypertrophy present in these patients are not precisely known. Recently, V. Simha et al.9 evaluated six patients with Dunnigan disease, observing an increased muscle fibre diameter suggestive of muscle hypertrophy. These authors concluded that muscle hypertrophy is not due to increased protein synthesis but rather due to a reduced protein catabolic pathway according to transcriptome data.
In summary, although it is reasonable to understand that not all physicians may have experience in the enormous number of Mendelian disorders that exist, all of them with very low prevalence, it is important that patients with singular phenotypes be referred to specialised referral units. To this end, our group has developed an application (Lipo-DDx®) to help physicians who are not experts in lipodystrophy syndromes to identify this type of rare disorders of adipose tissue.10
Institutional review board statementThis study was approved by the ethics review panel of the Red Gallega de Comités de Ética de la Investigación [Galician Network of Research Ethics Committees] (approval code 2017/477) and carried out according to the ethical guidelines of the Helsinki Declaration.
Informed consent statementAll subjects provided written informed consent for participation in the study and for the publication of their clinical, biochemical and molecular information.
Conflicts of interestD.A.-V. has received honorarium from Amryt for scientific consultancy, talks and CME. The rest of the authors declare no conflict of interest.
We are indebted to the patients of this study for their collaboration.
