


Los adenomas hipofisarios corticotropos clínicamente silentes constituyen un cuadro poco frecuente. Su curso clínico es variable; mientras que algunos siguen una evolución insidiosa, otros manifiestan un carácter agresivo, sobre todo durante las recidivas tumorales. El diagnóstico definitivo es histológico, dada la ausencia de datos clínicos y analíticos derivados del hipercortisolismo.
Clinically silent corticotroph adenomas are rare. The clinical course of these tumors varies: while some have an insidious course, others behave aggressively, especially during tumoral recurrence. Given the absence of clinical and biochemical features of hypercortisolism, the definitive diagnosis is histological.
Los adenomas hipofisarios corticotropos clínicamente silentes (corticotropinomas silentes) son tumores hipofisarios con inmunohistoquímica positiva a corticotropina que no muestran datos clínicos ni analíticos derivados del exceso de cortisol. La escasa incidencia de estos tumores dificulta el conocimiento de su evolución clínica. La mayoría de los corticotropinomas silentes son macroadenomas al diagnóstico, que se presentan clínicamente con síntomas secundarios a la compresión tumoral1,2.
Presentamos aquí los casos de 2 pacientes afectas de dicho cuadro, para comprender el abordaje diagnóstico-terapéutico y la evolución de estos tumores.
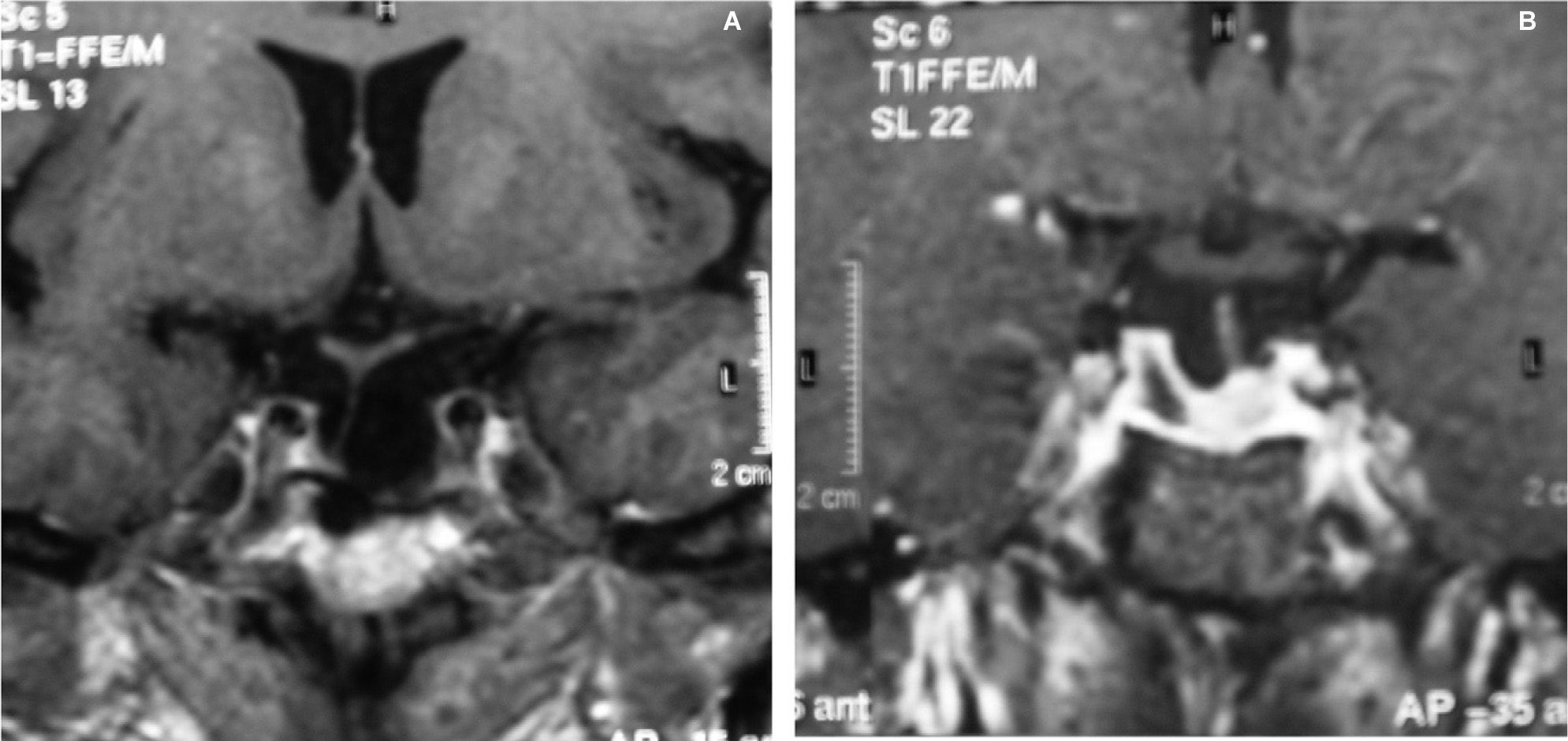
CASOS CLÍNICOSCaso 1Mujer de 60 años que consulta por cefalea holocraneal de carácter opresivo de 2 meses de evolución, fotofobia, alteraciones visuales, astenia y dolores osteoarticulares. En la exploración física, únicamente destacaba obesidad generalizada de grado 2 (índice de masa corporal [IMC] = 39,97) y hemianopsia bitemporal. La resonancia magnética (RM) hipofisaria objetivó una tumoración selar quística hiperdensa, de 15mm de diámetro, con centro hipodenso, extensión supraselar y compresión quiasmática. Las concentraciones hormonales basales mostraron un déficit de gonadotropinas (FSH, 3,12 U/l; LH, 0,21 U/l; normales: FSH, 2–13,8 U/l y LH, 2–13,8 U/l), hiperprolactinemia leve (47,6ng/ml; normal, 3,4-24,1ng/ml) y concentraciones de corticotropina elevadas (187,4pg/ml; normal, 9–52pg/ml) con concentraciones de cortisol plasmático (16,4μg/dl; normal, 8–25μg/dl) y de cortisol libre urinario (CLU) (42μg/24h; normal, 5–120μg/24h) dentro de la normalidad. No se observaron alteraciones de los ejes tiroideo y somatotropo. En cuanto a las pruebas de estímulo, se realizó una prueba de supresión nocturna con dexametasona (administración de 1mg de dexametasona a las 24h y determinación de cortisol y corticotropina a las 8h), y no se observó frenación del eje corticotropo (cortisol, 4,7μg/dl; corticotropina, 102pg/ml). Con el diagnóstico de macroadenoma hipofisario secretor de corticotropina con hipogonadismo secundario asociado, se practicó cirugía transesfenoidal que dejó un resto tumoral izquierdo. La anatomía patológica confirmó la presencia de un adenoma hipofisario, con inmunohistoquímica positiva al 100% para corticotropina, de distribución difusa, concordante con adenoma densamente granulado, y con coexpresión en un 10% de las células para prolactina, FSH y LH. Tras la cirugía, se produjo una mejoría de la alteración visual, con escotoma centrocecal y desaparición de la hemianopsia temporal inicial. El estudio de función hipofisaria evidenció, por un lado, el mantenimiento de concentraciones de corticotropina elevadas (127pg/ml) con concentraciones de cortisol dentro de la normalidad, tanto basales (15,6μg/dl) como tras estímulo con 250μg de corticotropina intravenosa (23,3-27,2μg/dl), y por otro, la persistencia de falta de supresión tras la administración nocturna de 1mg de dexametasona (corticotropina, 60,8pg/ml; cortisol, 2,6μg/dl). Además, presentaba un déficit de somatotropina (GH) (máximo, 1,9ng/ml tras estímulo con factor liberador de hormona del crecimiento [GHRH], con normalidad del resto de los ejes. Durante el seguimiento, se observó un aumento progresivo del resto tumoral en la porción lateral izquierda hipofisaria, y se inició tratamiento con radioterapia externa estereotáxica fraccionada, con una dosis total de 50Gy. Tras 21 meses, se evidencia una gran reducción del volumen tumoral, con persistencia de un resto quístico (1 × 0,5cm) (fig. 1A), mejoría de los síntomas (cefalea, astenia), e hipopituitarismo iatrogénico, con déficit de GH (IGF-I, 73ng/ml), gonadotropinas (FSH, 2,8 U/l; LH, 1,3 U/l) y déficit parcial de hormonas tiroideas (TSH, 2,12mU/l; tiroxina libre [T4L], 0,8ng/dl). En cuanto al eje corticotropo, mantiene concentraciones elevadas de corticotropina (71pg/ml), con cortisol en 14μg/dl y CLU en 15μg/24h, por lo que actualmente se encuentra cautelarmente en tratamiento con hidroaltesona.
Caso 2
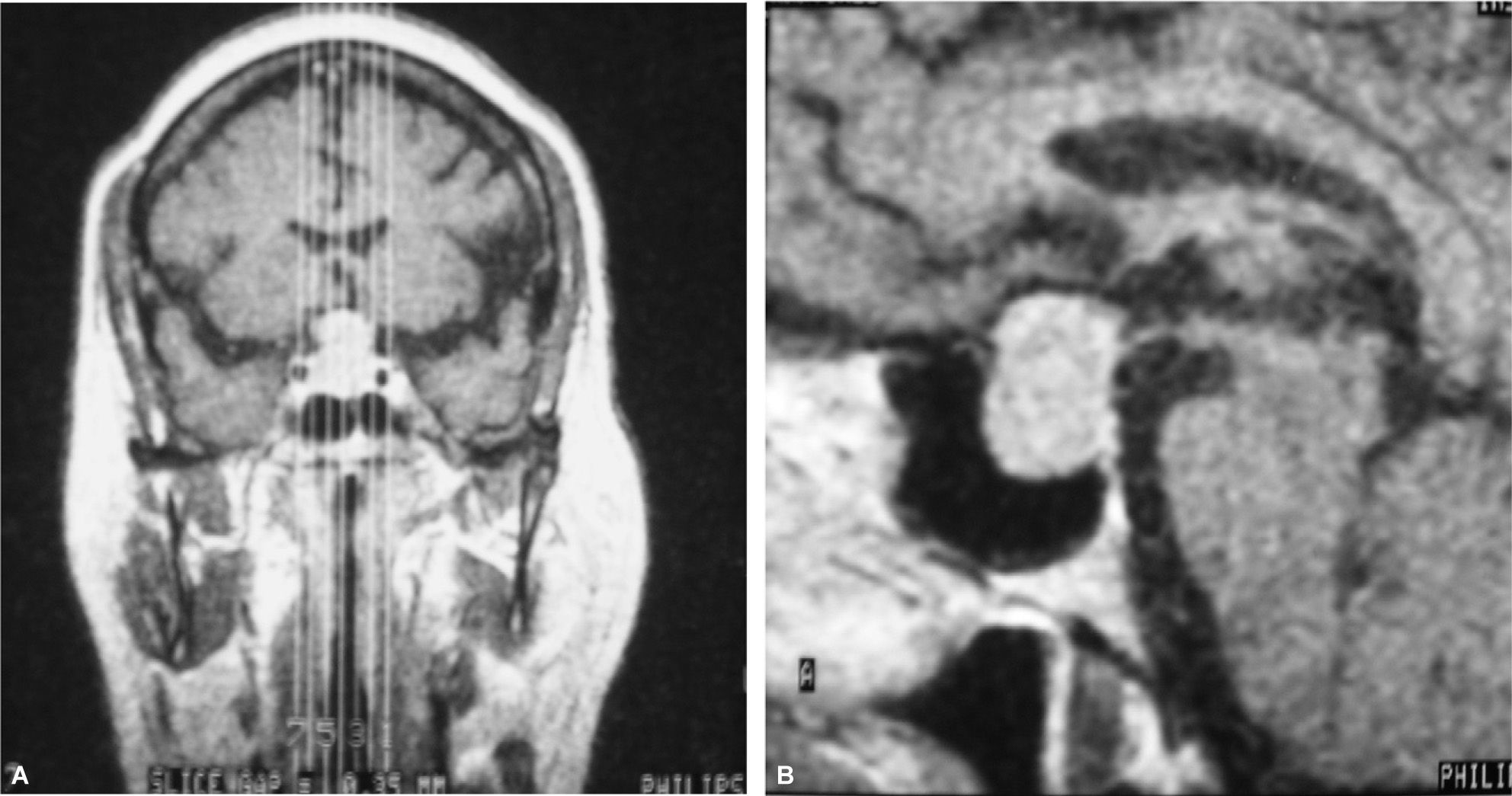
Mujer de 70 años con antecedentes de hipertensión arterial (HTA) en tratamiento con inhibidores de la enzima conversiva de angiotensina (IECA), cervicoartrosis y lesión calcificada occipital en seguimiento por neurocirugía. Refería cefalea, astenia y disminución de la agudeza visual de 2 años de evolución. En la exploración física destacaban una obesidad no troncular de grado 1 (IMC = 32,43) y estrías no violáceas abdominales. En la campimetría presentaba una reducción concéntrica de isopteras en ambos ojos que respetaba la región temporal inferior del ojo derecho. Se realizó una RM craneal, que puso de manifiesto una tumoración selar (2,2 × 1,5cm) con extensión a ambos senos cavernosos y seno esfenoidal y desplazamiento quiasmático (fig. 2). Las determinaciones hormonales basales mostraron hiperprolactinemia (69,5ng/ml), déficit de GH (basal, < 0,2ng/ml; IGF-I, 92ng/ml), con concentraciones plasmáticas de cortisol ligeramente elevadas (28,2μg/dl) y normalidad de las concentraciones de CLU (11μg/24h) y de los ejes tiroideo y gonadotropo. Se realizó resección transesfenoidal del tumor hipofisario. El informe anatomopatológico confirmó un adenoma hipofisario cromófobo, con una inmunohistoquímica altamente positiva para corticotropina y negativa para el resto de las hormonas adenohipofisarias. Dada la ausencia de signos, síntomas y bioquímica de hipercortisolismo asociado, el hallazgo es compatible con el diagnóstico de adenoma corticotropo hipofisario clínicamente silente. Tras la cirugía, la paciente presentó déficit somatotropo (GH basal < 0,2ng/ml; máximo tras GHRH, 1,7ng/ml; IGF-I, 97ng/ml), gonadotropo (FSH basal, 10,8 U/l; máximo tras GnRH, 14,6 U/l; LH basal, 2,5U/l; máximo tras GnRH, 7,9 U/l) y tiroideo (TSH, 2,5mU/l; máximo tras TRH, 14,1mU/l; T4L, 0,8ng/dl). El eje corticotropo se encontraba conservado, con concentraciones normales de cortisol (basal, 14,8μg/dl; 26,4μg/dl tras prueba de corticotropina), corticotropina (29,2pg/ml), CLU (14μg/24h), y supresión adecuada tras 1mg de dexametasona nocturna (cortisol, 1,2μg/dl). En RM seriadas, persiste un resto tumoral derecho (1,2 × 0,9 × 1,2cm) 6 años después de la cirugía (fig. 1B). Durante el seguimiento se produjo mejoría de los déficit visuales, con persistencia de astenia y cefalea.
DISCUSIÓN
Los adenomas hipofisarios corticotropos clínicamente silentes constituyen una entidad caracterizada por células tumorales hipofisarias inmunorreactivas a corticotropina y ausencia de datos clínicos y bioquímicos de hipercortisolismo. En la serie descrita por Horvath y Kovacs (de 300 casos), suponen un 5,7% de todos los adenomas hipofisarios intervenidos y un 43% de los adenomas de origen corticotropo.
Como se observa en los 2 casos presentados, se trata habitualmente de macroadenomas que cursan con signos y síntomas derivados del crecimiento y la compresión tumoral de estructuras vecinas. Los síntomas iniciales en el momento del diagnóstico suelen ser defectos visuales campimétricos (61%), cefalea (50%) e hipopituitarismo (26%).
Analíticamente, al igual que en los demás adenomas hipofisarios no funcionantes, podemos encontrar indicios de hipopituitarismo parcial o panhipopituitarismo secundarios a la compresión de la adenohipófisis por el gran tamaño tumoral3.
Las concentraciones de prolactina moderadamente elevadas previsiblemente se deban a la compresión del tallo hipofisario, aunque algunos autores apuntan la posibilidad de que se deba a la secreción de factores humorales paracrinos por el tumor4.
La concentración de corticotropina puede ser normal o moderadamente elevada. En el caso 1, se observa concentraciones elevadas de corticotropina circulante que persisten tras cirugía transesfenoidal y radioterapia, pero con concentraciones normales de cortisol y sin fenotipo cushingoide asociado. Se han establecido numerosas teorías para intentar explicar por qué el aumento de corticotropina no condiciona un hipercortisolismo secundario: aumento del catabolismo lisosómico, la existencia de un complejo de Golgi menos desarrollado que condicionaría un almacenamiento inadecuado de corticotropina en los gránulos neurosecretores y la secreción de hormona biológicamente inactiva1,5,6.
Con respecto a esto último, Reincke et al7 identificaron, en un paciente con corticotropinoma silente y aumento de concentraciones circulantes de corticotropina, dos máximos mediante cromatografía: el primero, correspondiente a la molécula de corticotropina esperable (1-39-ACTH), sólo suponía un 5% del total. El 95% restante estaba compuesto por corticotropina circulante de mayor peso molecular, a la que se atribuye la escasa actividad biológica. Yamakita et al8 posteriormente constataron similares resultados. El origen de esa corticotropina de mayor peso molecular (big ACTH) se encontraría en una alteración de su precursor (propiomelanocortina), hecho ya descrito y comprobado en pacientes con secreción ectópica de corticotropina7. La menor actividad biológica de dicha molécula explicaría por qué concentraciones elevadas de corticotropina no se correlacionan con el esperable incremento de las concentraciones de cortisol.
Existen tres subtipos de corticotropinomas silentes, clasificados mediante inmunohistoquímica y microscopio electrónico: el subtipo I es superponible morfológicamente a los adenomas corticotropos funcionantes; el subtipo 3 se asocia con un curso más agresivo9. En ninguno de los casos se evidencia la típica degeneración hialina de Crooke, presente en las células hipofisarias corticotropas no tumorales que rodean a las células tumorales en los pacientes con enfermedad de Cushing.
Clásicamente, se ha descrito un comportamiento agresivo de estos tumores1,2, con extensión supraselar (87%), apoplejía, infarto y cambios quísticos (61%). Takumi et al10 pusieron de manifiesto el caso de un paciente con apoplejía hipofisaria y síndrome del seno cavernoso. El análisis anatomopatológico e inmunohistoquímico demostró un adenoma hipofisario con células cosecretoras de corticotropina y somatotropina. Se trataba de un corticotropinoma silente, ya que, aunque las concentraciones de corticotropina y cortisol se encontraban marcadamente elevadas, probablemente por el estrés secundario a la enfermedad aguda, el paciente no presentaba rasgos clínicos atribuibles al hipercortisolismo. De cualquier forma, no es ésta la presentación habitual del cuadro, tal y como lo demuestra el curso indolente del tumor en nuestras 2 pacientes.
Los adenomas hipofisarios no funcionantes corticotropos presentan un elevado índice de recurrencia posquirúrgica6,7,10,11. En una serie reciente9, se comparó a 60 pacientes con adenomas hipofisarios no funcionantes no corticotropos y 28 pacientes con adenomas no funcionantes corticotropos. Ambos grupos presentaban un índice de recurrencia similar (32-33%), aunque las recurrencias de los adenomas corticotropos tenían un carácter más agresivo que las de los no corticotropos. Se observó que el grado de inmunorreactividad a la corticotropina y la extensión tumoral previa a la cirugía no se correlacionaban con la mayor o menor probabilidad de recurrencia posterior.
Por lo tanto, el abordaje inicial de estos tumores debe ser similar, con el empleo de técnicas quirúrgicas (transesfenoidal o transcraneal) como primera alternativa.
Después habría que llevar a cabo un estrecho seguimiento, con realización periódica de campimetría y RM hipofisaria, dados el alto índice de recurrencias y la posible mayor agresividad de los adenomas corticotropos. En caso de recidiva tumoral, se propone el uso de radioterapia como mejor alternativa terapéutica, sobre todo si se cumplen las siguientes características: resección quirúrgica incompleta, invasión parahipofisaria, significativa extensión supraselar tras la cirugía e indicios histológicos de malignidad9. En el caso de nuestras dos pacientes, se realizó en primer lugar cirugía hipofisaria transesfenoidal, con persistencia de resto tumoral en ambos casos. Dada la observación de crecimiento tumoral durante el seguimiento, se programó el inicio de radioterapia estereotáxica que, aunque a largo plazo demostró una disminución del tamaño de la masa selar, no condicionó una curación total del cuadro.
En cuanto a la posibilidad de tratamiento médico, hay datos suficientes para el uso de agonistas dopaminérgicos en los adenomas hipofisarios no funcionantes. En un estudio reciente12, se documentó una reducción tumoral significativa en un 60% y una mejoría de los síntomas atribuibles al tamaño tumoral en un 70-80% de los pacientes tras 1 año de tratamiento con cabergolina. Se observó una mayor respuesta en los tumores con una proporción más elevada de receptores dopaminérgicos D2, en particular con su isoforma corta. En casos aislados se ha descrito el uso de bromocriptina para prevenir la recurrencia de adenomas corticotropos agresivos10. Recientemente13, se ha publicado el primer caso de reducción tumoral de un adenoma corticotropo no funcionante recurrente tras tratamiento con cabergolina durante 2 años. Igualmente se demostró, mediante técnicas de hibridación in situ, que la proporción de receptores D2 hallados en el tumor era cercana a la encontrada en los prolactinomas usados como control. En cualquier caso, son necesarios más estudios que demuestren la eficacia terapéutica del uso de agonistas dopaminérgicos en los adenomas hipofisarios corticotropos no funcionantes.
Se han descrito casos aislados de corticotropinomas silentes que desarrollan enfermedad de Cushing tras años de evolución, lo que indica que la actividad biológica de los adenomas hipofisarios puede variar con el tiempo6,14. En nuestros casos, a pesar del prolongado tiempo de seguimiento (4 y 7 años, respectivamente), no se ha objetivado hasta ahora progresión de la enfermedad hacia la funcionalidad.
En conclusión, los adenomas hipofisarios corticotropos clínicamente silentes, son macroadenomas sin datos clínicos ni bioquímicos de hipercortisolismo, cuyo diagnóstico es fundamentalmente histopatológico, mediante el hallazgo de células tumorales hipofisarias inmunohistoquímicamente positivas a corticotropina. Recurren con frecuencia y de manera agresiva a pesar del tratamiento quirúrgico y radioterápico, por lo que es fundamental un estrecho seguimiento a largo plazo.
