


INTRODUCCIÓN
La diabetes mellitus es una de las patologías crónicas más frecuentes de la edad pediátrica. La diabetes es un síndrome que engloba alteraciones del metabolismo hidrocarbonado, proteico y lipídico, y es secundaria a una deficiente secreción o acción de la insulina. Se caracteriza por la existencia de concentraciones elevadas de glucosa en ayunas y postingesta. La diabetes no es una enfermedad única, sino que engloba a un grupo heterogéneo de alteraciones con distinto patrón genético, así como diferentes causas etiológicas y mecanismos fisiopatológicos.
En el síndrome diabético podemos distinguir 2 grandes grupos según el defecto esté, principalmente, en el déficit de secreción de insulina o sea debido a una defectuosa actuación de ésta. En el primer grupo, el déficit grave de la secreción de insulina puede ser secundario a varios mecanismos, entre los que cabe destacar: la destrucción autoinmunitaria de las células ß como en la diabetes tipo 11, defectos mitocondriales que interfieren en la generación de la energía intraislotes necesaria para la secreción de insulina como en la diabetes mitocondrial2,3, la agenesia pancreática como en el defecto homocigoto en el factor de transcripción del factor 1 promotor de la insulina (IPF-1)4; ser secundaria a la afectación de las células de los islotes de Langerhans como en la fibrosis quística de páncreas5,6, talasemia, cistinosis o en la afectación de las células ß por tóxicos como la l-asparraginasa, ciclosporina o tracrolimus7. También puede ser resultante de la pancreatectomía realizada en niños por hipoglucemias graves secundarias a hiperinsulinismo. Dentro de este grupo de diabetes por déficit de la secreción de insulina hay que incluir la diabetes familiar autosómica dominante debida a defectos genéticos en factores que intervienen en la función de la célula ß, la denominada diabetes tipo MODY (maturity onset diabetes of the young)8. De todas estas formas de diabetes insulinodependientes, la forma más frecuente en la infancia es la diabetes tipo 1 autoinmunitaria, que constituye en nuestro medio entre el 80 y el 90% de las diabetes y, en segundo lugar, la diabetes tipo MODY, que representa alrededor del 5%.
El otro gran grupo, en el que existe principalmente una resistencia a la acción de la insulina (aunque con afectación concomitante de la función de la célula ß), se encuentra la diabetes tipo 2. Clásicamente, la diabetes tipo 2 (DM2) se ha considerado una enfermedad exclusiva de los adultos. Sin embargo, en la última década ha habido un incremento llamativo de su incidencia en la edad pediátrica, sobre todo en adolescentes9,10. En algunas poblaciones estos incrementos han sido espectaculares y se han considerado epidémicos. Este incremento ha sido paralelo al aumento en la prevalencia de la obesidad en la infancia.
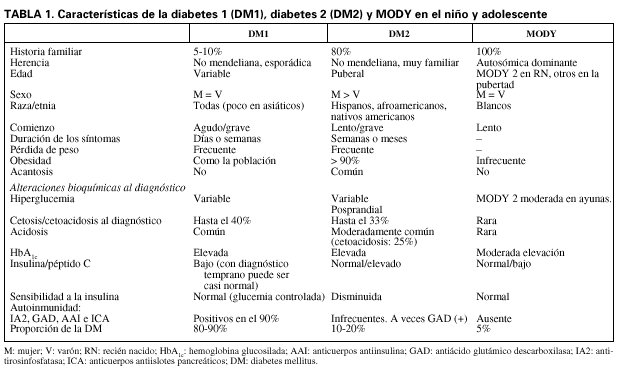
Por todo ello, ante el diagnóstico de diabetes en el niño, no debemos pensar sólo en la diabetes tipo 1, aunque ésta sea la más frecuente, sino que hay que descartar los otros tipos de diabetes. Esto es muy importante, ya que puede implicar actitudes terapéuticas diferentes. En esta revisión trataremos de analizar los aspectos que nos van a ayudar a hacer un diagnóstico correcto, para lo cual vamos a hacer hincapié en la epidemiología de los distintos tipos de diabetes, sus diferentes características clínicas y los distintos tipos de marcadores genéticos y bioquímicos que las diferencian (tabla 1).

DIABETES TIPO 1
En la diabetes tipo 1 (DM1) autoinmunitaria, tanto los factores genéticos como los ambientales influyen en el desarrollo de la enfermedad. Los primeros contribuyen al 70-75% de la susceptibilidad a este tipo de diabetes y los segundos parecen facilitar o desencadenar el proceso que lleva a la destrucción de las células ß al comienzo de la enfermedad. Este tipo de diabetes tiene un largo período preclínico. La existencia de autoanticuerpos antiinsulina, antiislotes pancreáticos, antiácido glutámico descarboxilasa y antitirosinfosfatasa indica la presencia del proceso autoinmunitario, y pueden estar presente años antes del diagnóstico de la enfermedad11.
La incidencia de la DM1 en la infancia se ha incrementado globalmente en los últimos 50 años en todas las poblaciones, tanto en las de baja como alta incidencia12,13. Hay una amplia variación de la incidencia de DM1 en la edad pediátrica en los diferentes países, así como entre y dentro de los distintos grupos étnicos, lo que indica la importancia de los factores ambientales en su etiología. Existe una gran variabilidad geográfica, de modo que la incidencia es más alta en los países del norte de Europa. La mayor incidencia se da en Finlandia, donde llega a afectar a 50 por cada 100.000 niños menores de 15 años14. Entre los países con una incidencia más baja está China, con 0,1 por 100.000. Nuestro país, aunque también con considerables diferencias geográficas, se encuentra entre los países de incidencia media, oscilando en las distintas zonas; en la Comunidad Autónoma de Madrid es de 14,4 por 100.000. Una de las zonas con una incidencia más alta en nuestro país es la de Málaga15. El pico de mayor incidencia de DM1 está entre los 10 y 14 años, coincidiendo con el período puberal, aunque se está observando un mayor incremento en el diagnóstico de diabetes en los niños menores de 5 años en casi todos los países16,17.
La DM1 se hereda ligada al complejo principal de histocompatibilidad, en el que cabe destacar como los de mayor riesgo DR3/4 y DQB0302/DQB0201.
Entre los aspectos clínicos a destacar encontramos que los pacientes con DM1 autoinmunitaria no tienen, en general, sobrepeso (aunque en la actualidad el 24% de los niños estadounidenses con DM1 sí lo presentan), manifiestan pérdida de peso reciente, poliuria y polidipsia generalmente de corta duración y, a menudo, cetosis. El 30-40% comienza con cetoacidosis. Después de la estabilización inicial pueden presentar un período de remisión con menores necesidades de insulina. El 5% tiene un familiar de primer o segundo grado con DM1. Estas características, junto a la presencia de marcadores de autoinmunidad, concentraciones bajas de insulina y péptido C, son la base del diagnóstico de este tipo de diabetes.
DIABETES TIPO 2
Hasta hace poco, la DM1 mediada por mecanismos autoinmunitarios era el único tipo de diabetes que se consideraba prevalente entre los niños, mientras que se estimaba que sólo el 1-2% de los niños presentaban DM2 y otras formas raras de diabetes. Informes recientes indican que entre el 8 y el 45% de los niños con DM de EE.UU. presentan una diabetes no mediada por mecanismos autoinmunitarios, aunque esta incidencia no se ve en nuestro país, donde no pasa de 2 al 3%. Debido a que el reconocimiento de la existencia de DM2 en la población pediátrica es relativamente nuevo, los datos de su incidencia y prevalencia son incompletos y no hay grandes series de pacientes estudiados18-22.
La incidencia y prevalencia estimadas de la DM2 en la edad pediátrica continúan aumentando y varían según la edad y etnias consideradas. Esta enfermedad se expresa con mayor frecuencia en etnias concretas, entre ellas los indios americanos, afroamericanos, hispanos y asiáticos-americanos. Pero incluso la incidencia ha aumentado entre los caucásicos de EE.UU.23. Es posible que exista una infraestimación de la frecuencia de la DM2, ya que muchos casos pueden no estar diagnosticados porque en el momento del diagnóstico a veces es difícil distinguir entre DM1 y DM2, sobre todo en los adolescentes. Un estudio realizado en los niños y adolescentes del Registro de Diabetes de Chicago demostró que el 25% de ellos estaban erróneamente diagnosticados como diabéticos tipo 1 cuando en realidad tenían DM224.
Entre las causas del incremento de su incidencia está el aumento de prevalencia de la obesidad, que está adquiriendo proporciones epidémicas en los adolescentes de EE.UU., donde hasta el 20% la presenta25. En España la incidencia de la obesidad oscila entre el 6 y el 10%26,27, y en general los casos son menos graves. En este incremento de la obesidad intervienen el aumento de la ingesta calórica y lipídica y la disminución de la actividad física.
También el bajo peso al nacer y la existencia de diabetes en la madre durante el embarazo se han involucrado en el incremento de la incidencia de DM2. Un estudio en indios pima ha demostrado que el peso al nacimiento por debajo de 2,5 kg o por encima de 4,5 kg se asoció a un riesgo incrementado de presentar DM2 en la adolescencia. La relación entre diabetes de la madre durante el embarazo y la obesidad e incremento del riesgo de diabetes en sus descendientes también se ha documentado en los indios pima28. Los mecanismos fisiopatológicos que contribuyen a ello no se conocen bien. Parece que el ambiente intrauterino de una madre diabética puede ser determinante en el desarrollo posterior de la diabetes (causante de resistencia insulínica y de alteración de función de la célula ß) junto a la influencia de los factores genéticos.
El incremento de la DM2 en la infancia suele ocurrir en poblaciones con alta frecuencia de DM2 entre los adultos. No hay datos de prevalencia de la DM2 en España. En nuestra experiencia con 300 niños y adolescentes seguidos por diabetes, la DM2 supone el 2%.
Pese a la amplia experiencia y conocimientos sobre la epidemiología, fisiopatología y tratamiento médico de la DM2 en adultos, sabemos poco sobre esta enfermedad en los niños y adolescentes. La Asociación Americana de Diabetes convocó una conferencia consenso sobre DM2 en la edad pediátrica en agosto-septiembre de 1999, con un panel de 8 expertos en diabetes pediátrica, representantes de la Academia Americana de Pediatría y otros organismos de adultos24. Este grupo desarrolló una postura de consenso en los diferentes aspectos de la DM2 en la infancia.
La DM2 es un trastorno metabólico complejo de etiología heterogénea. En el desarrollo de la enfermedad intervienen factores genéticos (poligénicos) y ambientales (obesidad, vida sedentaria, dieta rica en grasas y pobre en fibras) que influyen en la expresión de la enfermedad29. El papel determinante de los factores genéticos salió a la luz al considerar las diferencias de prevalencia de la DM2 en los distintos grupos raciales, pero el reciente incremento de prevalencia de esta enfermedad respalda la importancia de los factores medioambientales en su expresión.
Se sabe que en la fisiopatología de la DM2 están involucrados defectos de la acción de la insulina (resistencia insulínica) y alteración en la secreción de insulina que llevan a hiperglucemia crónica. Con frecuencia los niños con DM2 tienen signos de insulinorresistencia, como es la presencia de acantosis nigricans y la esteatohepatitis.
La pubertad desempeña un papel clave en la manifestación clínica de la DM2 en el adolescente debido a la insulinorresistencia que existe fisiológicamente durante ese período, en parte inducida por el aumento de la hormona del crecimiento coincidiendo con el estirón puberal (en el individuo normal este incremento queda compensado por el aumento de la secreción de la insulina).
Hay diferencias raciales en cuanto a la sensibilidad a la insulina también en la infancia (los afroamericanos adolescentes tienen una sensibilidad a la insulina un 30% más baja que los adolescentes de raza blanca). Estos datos indican que, en los niños con predisposición genética a la insulinorresistencia, la presencia de moduladores ambientales puede aumentar el riesgo de desarrollar DM2 y determinar la expresión de la enfermedad durante estadios fisiológicos (pubertad) o patológicos (obesidad) de insulinorresistencia30.
En un estudio realizado por nuestro grupo en 159 pacientes obesos de entre 4 y 18 años de edad, encontramos que sólo 2 pacientes presentaban alteración de la tolerancia a la glucosa y ninguno diabetes31. El 38% de los pacientes tenía datos de insulinorresistencia (HOMAR > 2,5) con correlación positiva entre el HOMAR y las concentraciones de glucosa a las 2 h de la sobrecarga oral de glucosa, el índice insulinogénico, los triglicéridos, la cifra de alaninaaminotransferasa, el ácido úrico e índice de masa corporal, y una correlación negativa con los valores de colesterol ligado a lipoproteínas de alta densidad (datos de síndrome metabólico). Once pacientes tenían una cifra de alaninaaminotransferasa mayor de 40 U/l y, de ellos, el 72% tenía en la ecografía esteatohepatitis no alcohólica, con hiperinsulinismo en el 75% de los casos. En un paciente la biopsia hepática evidenció esteatosis y fibrosis confluente. Nuestros resultados son muy diferentes de los encontrados en los estudios en niños y adolescentes realizados en EE.UU., en los cuales la incidencia de diabetes es alta; esto puede deberse al mayor grado de obesidad que presenta su población, además de las influencias genéticas.
Clínica
La DM2 predomina en las niñas. El comienzo suele ser insidioso, con pocos síntomas, lo que hace que con frecuencia el diagnóstico sea tardío. Las descompensaciones agudas pueden ocurrir, y hasta el 25% de los pacientes presentan cetosis en el momento del diagnóstico. La pérdida de peso es escasa o nula32,33.
La edad media de diagnóstico es entre los 12 y 14 años; coincide con la insulinorresistencia relativa que existe en la pubertad. La mayoría de los pacientes presenta sobrepeso, con un índice de masa corporal superior al percentil 95 para la edad y el sexo. Es muy frecuente la historia familiar de DM2; el 45-80% tiene un progenitor con DM2, y un 74-100%, algún familiar de primera o segunda generación con DM2 (éste puede no estar diagnosticado en ese momento). La presencia de acantosis nigricans es casi constante (90%) y frecuente el síndrome de ovario poliquístico.
Diagnóstico
Para el diagnóstico de la DM2, además de los signos clínicos y síntomas, se precisa la evaluación de laboratorio y de la evolución. Se usan los criterios emitidos en 1997 por el Comité de Expertos de la Asociación Americana de Diabetes. De acuerdo con este comité, el término DM2 debe usarse para aquellos individuos que: a) presentan insulinorresistencia y déficit relativo de insulina; b) no necesitan insulina para sobrevivir, al menos al inicio; c) no presentan destrucción autoinmunitaria de la célula ß, y d) no hay evidencia de otras causas de diabetes (defectos genéticos de la acción de la insulina, enfermedades del páncreas exocrino, diabetes inducida por fármacos, etc.).
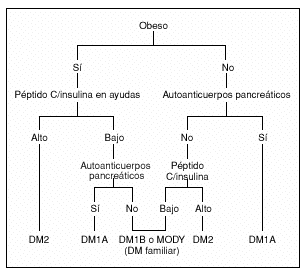
Los datos a favor de la DM2 son: obesidad (índice de masa corporal elevado), existencia de acantosis nigricans, historia familiar de DM2, etnicidad (nativos americanos, afroamericanos, americanos de origen hispano), péptido C normal con ausencia de autoanticuerpos pancreáticos, tratamiento con dieta, ejercicio y antidiabéticos orales (fig. 1).

Fig. 1. Niño o adolescente con hiperglucemia24. DM: diabetes mellitus.

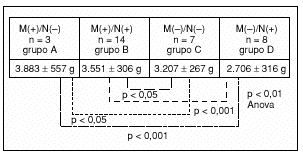
Fig. 2. Influencia de la presencia de mutación de la glucocinasa materna (M+) y del feto (N+) en el peso al nacimiento43.
En el caso de los niños, el diagnóstico es difícil porque en ellos la DM2 puede presentarse en cetoacidosis, aunque con menos frecuencia que en los diabéticos tipo 1. Los autoanticuerpos antipancreáticos pueden ser negativos en un porcentaje pequeño de diabéticos tipo 1 y algunos pacientes con DM2 presentan anticuerpos antiácido glutámico descarboxilasa.
DIABETES TIPO MODY
La diabetes tipo MODY, que comprende un grupo de enfermedades distintas, pertenece a un subtipo monogénico de diabetes mellitus, caracterizado por un comienzo temprano, habitualmente antes de los 25 años, herencia autosómica dominante y disfunción primaria de la célula ß pancreática. El MODY supone del 1 al 5% de todos los casos de diabetes en los países industrializados.
La diabetes tipo MODY es genéticamente heterogénea y resulta de mutaciones en estado heterocigoto en, al menos, 6 genes diferentes34-36. Uno de estos genes codifica la enzima glucolítica, la glucocinasa (GK; MODY 2), que interviene como sensor de la glucosa en la regulación de la secreción de la insulina. Los otros 5 genes implicados codifican factores de transcripción de la célula ß: el IPF-1 (MODY 4), los genes de los factores nucleares hepáticos (HNF) 1* (MODY 3), 4* (MODY 1) y 1ß (MODY 5), que se expresan en el hígado y en los islotes pancreáticos, y el factor 1 de diferenciación neurogénica (Neuro-D1, también denominado ß2; MODY 6), que interviene en el desarrollo pancreático y en la transcripción del gen de la insulina.
En los últimos años, el descubrimiento de genes relacionados con diversos tipos de diabetes monogénica ha permitido una mejor comprensión de la relación fenotipo/genotipo en este tipo de diabetes, así como un avance en el conocimiento de la homeostasis de la glucosa y del papel que diversos factores de transcripción desempeñan en el desarrollo y en la función de la célula ß pancreática.
Factores no genéticos pueden afectar a la edad de comienzo y a la gravedad de la hiperglucemia. Los sujetos con distintos tipos de MODY tienen características clínicas diferentes y anomalías específicas en la secreción de insulina estimulada por la glucosa8.
Alteraciones del gen de la glucocinasa (MODY 2)
Las mutaciones del gen de la GK fueron los primeros defectos genéticos descritos en un gen involucrado en el metabolismo de la glucosa e implicado en la predisposición genética a la diabetes. La GK se expresa en concentraciones elevadas en la célula ß. Los sujetos con mutaciones de la GK tienen un umbral más alto de glucosa para la estimulación de la secreción de insulina que los individuos normales. Este umbral en el sujeto normal está próximo a los 5 mmol/l (90 mg/dl) y la GK actúa en la célula ß como sensor37. En los defectos de GK el umbral de glucosa se sitúa entre 6 y 7 mM (108-126 mg/dl), lo que lleva a un incremento de la glucemia basal y posprandial. Sin embargo, mecanismos compensatorios que operan in vivo, como es el efecto primador de la glucosa en la secreción de la insulina, limitan la gravedad del defecto secretorio de insulina y, por tanto, el grado de hiperglucemia observado en el curso de la enfermedad.
Al tiempo, la alteración de la GK hepática lleva a una disminución de la acumulación neta de glucógeno hepático y a un aumento de la neoglucogénesis después de las comidas, lo que exacerba la hiperglucemia posprandial38.
Las mutaciones heterocigotas del gen de la GK que dan lugar a un déficit parcial de la enzima se asocian al MODY 2. Hasta la actualidad se han descrito más de 130 mutaciones diferentes que codifican una proteína con la actividad enzimática modificada39. El MODY 2 se ha encontrado en personas de todas las razas y grupos étnicos. Las alteraciones metabólicas del MODY 2 están presentes desde el nacimiento, suelen ser leves y en general no precisan tratamiento medicamentoso; sólo el 2% requiere insulina8. La incidencia de complicaciones crónicas es muy baja. Cerca del 50% de las mujeres portadoras de la mutación en el gen de la GK tienen diabetes gestacional40.
Estudios realizados por Hattersley et al41,42, posteriormente corroborados por nuestro grupo43 (fig. 2), mostraron que el defecto genético del feto puede ser el causante del bajo peso al nacimiento en estos niños cuando la madre no porta la mutación (la reducción es de alrededor de 500 g), lo que demuestra la influencia que los cambios en la secreción de insulina fetal pueden tener en el crecimiento intrauterino. En el caso de que la madre tenga un MODY 2, la hiperglucemia materna hace que se sobrepase el umbral glucémico para la liberación de la insulina y que disminuya la insulinopenia prenatal.
En nuestro estudio, realizado en 22 familias españolas, encontramos que el MODY 2 es la forma más frecuente de MODY en la población pediátrica43. Estos resultados son semejantes a los encontrados por los italianos44 y france ses45, y difieren de los estudios ingleses y nórdicos, que encuentran que el MODY 3 es el predominante en sus poblaciones35,46. La discrepancia entre estos resultados puede deberse a que la población estudiada era distinta: predominantemente infantil en Francia, Italia y en nuestro estudio, y adulta en el estudio de Costa et al47 en Cataluña y en el resto de Europa.
Las alteraciones homocigotas de la GK se han implicado en 2 casos de diabetes neonatal permanente48.
Diabetes debida a alteraciones de los factores de transcripción
El MODY 3 es la forma más frecuente de diabetes tipo MODY en la mayoría de los países. En el Reino Unido supone el 63%, en Dinamarca el 60%, en Alemania el 36% y en España el 35% en el estudio realizado por Costa et al47 en población adulta, pero supone sólo el 18,18% en nuestra experiencia en población pediátrica43 y el 21% en Francia39,49. Hasta la actualidad se han identificado más de 120 mutaciones en personas de todas las razas y grupos étnicos8.
La mutación del gen HNF-1* lleva a una disfunción progresiva de la célula ß, que se caracteriza inicialmente por un fallo para incrementar la secreción de insulina ante las elevaciones de la glucemia. Con el tiempo, esta alteración progresa y se reduce globalmente, lo que lleva a una marcada hiperglucemia39. La hiperglucemia mantenida hace que estos pacientes tengan un riesgo alto de presentar complicaciones microvasculares si no se controlan de manera adecuada. Los pacientes con este tipo de diabetes son muy sensibles a la insulina y a las sulfonilureas, lo que hay que tener en cuenta al plantearse su tratamiento.
El defecto de la célula ß es progresivo. En experimentos fisiológicos se ha observado que aquellas personas que heredan la mutación HNF-1* y no son todavía diabéticas muestran concentraciones adecuadas de glucemia en ayunas pero son incapaces de incrementar la secreción de insulina de acuerdo con los valores de glucosa cuando éstos exceden los 8 mmo/l (144 mg/dl). En estos pacientes existe una considerable variación en el fenotipo dentro de las familias afectadas y en las distintas familias. No se ha encontrado una clara correlación fenotipo/genotipo. El fenotipo clínico de MODY 3 se parece, en parte, a la clásica DM2 en su historia natural, con pacientes que progresan rápidamente desde la alteración de la tolerancia a la glucosa a diabetes manifiesta, con un deterioro progresivo de la secreción de insulina.
Los sujetos con mutaciones en el gen HNF-1* presentan una respuesta disminuida de la secreción de insulina cuando los valores de glucemia exceden de 8 mmol/l (145 mg/dl), con un déficit grave de insulina e insulinorresistencia que parece ser secundaria a la hiperglucemia. Habitualmente, el MODY 3 se asocia con baja prevalencia de obesidad, dislipemia e hipertensión arterial.
La hiperglucemia, generalmente, se desarrolla después de la pubertad o en los primeros años de la edad adulta. Tiene una gran penetrancia; el 78% de los pacientes están afectos a los 35 años, con glucemias en ayunas superiores a 140 mg/dl. En la adolescencia y al inicio de la edad adulta pueden mostrar sólo una mínima elevación de la glucemia en ayunas, pero una respuesta diabética a la sobrecarga oral de glucosa50,51. Estos pacientes presentan con mucha frecuencia síntomas osmóticos en el momento del diagnóstico; esto parece reflejar el efecto específico de las mutaciones en HNF-1*, que reducen el umbral renal para la glucosa en el tubo renal proximal; se acompaña en general de aminoaciduria52.
Las mutaciones heterocigotas en el gen del HNF-4* son las causantes del MODY 153. Los mecanismos fisiopatológicos del MODY 1 y MODY 3 son muy similares debido a que el HNF-4* regula al HNF-1*.
En el MODY 1, las mutaciones genéticas se asocian con una incapacidad para incrementar la secreción de insulina cuando la concentración de glucemia es superior a 7-8 mmol/l (126-144 mg/dl), y el efecto normal de debado de la glucosa sobre la secreción de insulina se pierde. A pesar de tener similares elevaciones de la glucemia en ayunas que en el MODY 2, los valores de glucemia a las 2 h de una sobrecarga oral de glucosa son significativamente más elevados. La hiperglucemia del MODY 1 y del MODY 3 tiende a incrementarse con el tiempo de evolución, lo que lleva a la necesidad de tratamiento con antidiabéticos orales o insulina en una proporción importante de pacientes (del 30 al 40% requieren insulina). Estas formas de MODY se asocian con una disminución progresiva de la secreción de insulina (ésta disminuye del 1 al 4% por año)8. Estas alteraciones son evidentes en sujetos prediabéticos que tienen mutaciones del HNF-4*con concentraciones normales de glucosa, lo que indicaría que el defecto de la célula ß es un defec to patogénico primario en el síndrome diabético de estos su jetos.
El MODY 1 es muy poco frecuente. Hasta la actualidad sólo se han encontrado 13 familias en el mundo con MODY 18. En nuestro estudio realizado en 22 familias con MODY, hemos encontrado el primer caso de MODY 1 en España, con una mutación no descrita previamente43. Este tipo de diabetes tiene un riesgo importante de complicaciones crónicas si no se mantiene la euglucemia.
Las mutaciones del gen que codifica el IPF-1 es una causa rara de MODY (MODY 4). El conocimiento de esta forma de diabetes se basó en el estudio de una familia4,54. El caso índice era un niño con agenesia pancreática y diabetes neonatal permanente. El niño era homocigoto para una mutación del IPF y sus padres, heterocigotos, presentaban fenotipo de diabetes tipo MODY con múltiples familiares afectados. En la familia la expresión de la diabetes fue variable y las edades de presentación eran más tardías que en los otros tipos de MODY.
Mutaciones heterocigotas en el factor de transcripción HNF-1ßen el humano llevan al fenotipo del MODY 5, forma muy poco frecuente de MODY, que incluye diabetes, enfermedad renal temprana y progresiva no relacionada con la diabetes y malformaciones genitales.
En conclusión, en la primera infancia sólo se manifiesta el MODY 2 y en la pubertad se pueden expresar el MODY 3 y MODY 1. Los otros tipos de MODY suelen ser de expresión más tardía y son muy poco frecuentes. Para el diagnóstico de diabetes tipo MODY lo más importante es una buena historia a fin de recoger el patrón familiar de herencia autosómica dominante. Hay que tener en cuenta que estos pacientes no son obesos y presentan, en general, concentraciones bajas de insulina en relación con la glucemia, lo que les diferencia de los casos de DM2. En ellos no existen marcadores de autoinmunidad pancreática.
