


INTRODUCCIÓN
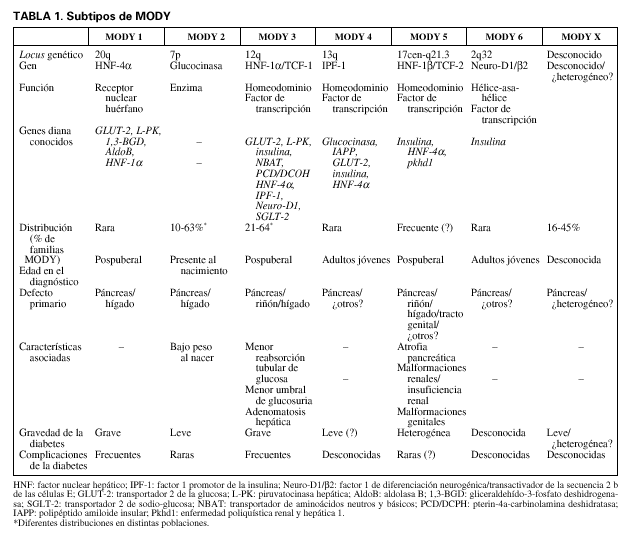
Con el término MODY, acrónimo de maturity onset diabetes of the young, se designa un grupo de afecciones caracterizadas por hiperglucemia no cetósica familiar con herencia autosómica dominante. La hiperglucemia en los sujetos afectados de MODY suele aparecer en niños, adolescentes o adultos jóvenes y se asocia con defectos primarios de la secreción de insulina. Las mutaciones heterocigóticas en 6 genes provocan la mayoría de los casos de MODY (tabla 1). Estos genes codifican la enzima glucocinasa (subtipo MODY 2)1 y los siguientes factores de transcripción: factor nuclear hepático 4* (HNF-4*; MODY 1)2, factor nuclear hepático 1* (HNF-1*; MODY 3)3, factor 1 promotor de la insulina (IPF-1; MODY 4)4, factor nuclear hepático 1ß (HNF-1ß; MODY 5)5 y factor 1 de diferenciación neurogénica (Neuro-D1, también denominado ß2; MODY 6)6. Probablemente hay además otros genes MODY, dado que existen familias en las que una diabetes clínicamente compatible con MODY no se cosegrega con marcadores íntimamente vinculados a los locus MODY conocidos7,8. Aunque no se dispone de estudios epidemiológicos definitivos, se supone que los diversos tipos de MODY pueden ser responsables del 2-5% de los casos de diabetes no insulinodependiente9.

Se ha observado que la prevalencia relativa de los diferentes subtipos de MODY varía considerablemente en los estudios de familias con MODY de distintas poblaciones7,10-13. MODY 2 representa del 8 al 63% de los casos, y MODY 3, del 21 al 64%. Los otros subtipos MODY son trastornos raros que se han descrito únicamente en unas pocas familias, mientras que otros locus desconocidos, únicos o múltiples (MODY X), representan del 16 al 45% de los casos de MODY. Esta diferencia de resultados puede deberse a divergencias en el trasfondo genético de las poblaciones, o bien, al menos en parte, a un sesgo de determinación en el reclutamiento de las familias.
Los subtipos genéticos de MODY presentan una notable heterogeneidad clínica por lo que se refiere a su modo de presentación, gravedad del déficit insulínico, evolución, grado de hiperglucemia, frecuencia de complicaciones microvasculares y existencia de otras anomalías asociadas con la diabetes (tabla 1). En esta revisión se debatirá la heterogeneidad clínica y metabólica asociada con la he terogeneidad genética de MODY y, cuando sea posible, las consecuencias para el tratamiento de la diabetes. El debate se centrará esencialmente sobre MODY 2 y MO DY 3, que son, con mucho, las formas más prevalentes de MODY.
MODY 2
Las mutaciones heterocigóticas en el gen de la glucocinasa son una causa común de MODY. Se han hallado más de 280 mutaciones distintas en sujetos de múltiples procedencias raciales y étnicas diferentes14,15 (Bellanné-Chantelot et al, resultados no publicados). Es un hecho notable que las mutaciones homocigóticas o heterocigóticas dobles en el gen de la glucocinasa den lugar a un fenotipo diferente, lo que conduce a una diabetes mellitus neonatal permanente16, aunque no son al parecer una causa frecuente de este tipo de diabetes17.
Fisiopatología de las mutaciones en el gen de la glucocinasa
La glucocinasa cataliza la fosforilación de la glucosa en posición 6 y desempeña un papel crucial en la regulación e integración del metabolismo de la glucosa en las células ß pancreáticas y en los hepatocitos. En las células ß pancreáticas, el metabolismo de la glucosa y la secreción de insulina dependen en gran medida de la actividad enzimática de la glucocinasa, que actúa como un sensor de la glucosa. En los estudios de expresión se ha observado que las actividades enzimáticas de los mutantes de glucocinasa detectadas en las familias MODY 2 estaban alteradas18,19, lo que daba lugar a que disminuyera el flujo glucolítico en las células ß pancreáticas. Este defecto se traduce in vivo en un trastorno en la capacidad para percibir la glucosa, lo que da lugar a un aumento en el umbral de glucemia que desencadena la secreción de insulina, desde una concentración basal normal de 5 mM a unos 6-7 mM20,21, así como a una desviación a la derecha en la curva de dosis-respuesta que relaciona la secreción de insulina con las concentraciones de glucosa21. Al comparar las tasas de secreción de insulina frente a una amplia gama de concentraciones de glucosa, se demostró que los sujetos con MODY 2 con déficit de la glucocinasa presentan una reducción media del 60% en la secreción de insulina ante una determinada cifra de glucosa21. Como hecho de interés, los valores de insulina en los sujetos MODY 2 suelen ser normales durante todo el día, a expensas de la hiperglucemia21. La liberación de insulina en respuesta a otros secretagogos, como la arginina, suele hallarse bien conservada o sólo moderadamente disminuida22, lo cual indicaría que este defecto secretorio estaría relacionado con una relativa ceguera a la glucosa por parte de las células ß pancreáticas. La adaptación fisiológica en el seno de dichas células limita la gravedad del defecto de secreción insulínica. Los estudios realizados en modelos animales muestran que este mecanismo compensador se halla relacionado con una mayor expresión, inducida por la glucosa, del único alelo natural del gen de la glucocinasa23.
Aunque el principal mecanismo fisiopatológico de la hiperglucemia en los pacientes con mutaciones en el gen de la glucocinasa es la disfunción de las células ß, debe señalarse que las anomalías en el metabolismo hepático de la glucosa contribuyen a la patogenia de la hiperglucemia en la MODY 2. En el hepatocito la glucocinasa interviene en el primer paso del almacenamiento de la glucosa en forma de glucógeno. En los pacientes con MODY 2, la síntesis hepática de glucógeno está reducida, y la neoglucogénesis, aumentada24. El aumento posprandial de la tasa relativa de neoglucogénesis es probablemente un importante factor que contribuye a la moderada hiperglucemia posprandial que se observa en los sujetos con MODY 225. En apoyo de ello, se ha observado una supresión anormal de la producción hepática de glucosa ante concentraciones fisiológicas de insulina en sujetos MODY 2 durante un pinzamiento euglucémico26. Además, los sujetos MODY 2 presentan una disminución del ciclaje hepático de glucosa, así como una producción endógena de glucosa anormalmente elevada en relación con las concentraciones plasmáticas de glucosa, y también una supresión amortiguada tras la administración oral de glucosa27. En conjunto, estas observaciones inducen a pensar que el hígado desempeña un papel en la fisiopatología de la hiperglucemia relacionada con el déficit de glucocinasa, aunque probablemente dicho papel es inferior al que tienen las células ß pancreáticas. Aunque algunos sujetos con MODY 2 pueden presentar además resistencia periférica a la insulina, la disminución de la sensibilidad a ésta probablemente no esté relacionada con las mutaciones de la glucocinasa26.
Características clínicas de MODY 2
A pesar de las considerables diferencias en la actividad enzimática in vitro de las distintas mutaciones de la glucocinasa18,19, el fenotipo es notablemente similar en todos los pacientes con MODY 225. Los pacientes con mutaciones de la glucocinasa presentan durante toda su vida una ligera hiperglucemia en ayunas (por lo común, de 5,5 a 8,0 mM), que se regula a esta concentración más elevada. En una serie de 260 sujetos de 42 familias francesas afectadas de MODY 2, correspondientes a 36 mutaciones diferentes, la glucemia en ayunas era de 7 mM por término medio1. En esa serie, menos del 50% de los sujetos portadores de una mutación de la glucocinasa cumplían los criterios para la diabetes, según la definición actual basada en la glucemia en ayunas (>= 7 mM). En un estudio multicéntrico europeo realizado en más de 250 pacientes, no se halló ningún sujeto con una glucemia en ayunas uniformemente inferior a 5,5 mM25. La hiperglucemia se halla presente desde el nacimiento y puede diagnosticarse a cualquier edad a partir del período neonatal1. La edad del diagnóstico es simplemente aquella en que se comprueba por primera vez la hiperglucemia. Es raro que los pacientes presenten hiperglucemia sintomática y la mayoría de ellos se diagnostican casualmente o en una prueba de detección familiar1. Es típico que aumente poco la glucemia después de las comidas o de una sobrecarga oral de glucosa. En la prueba de tolerancia oral a la glucosa (PTOG), el incremento a las 2 h respecto a los valores en ayunas suele ser inferior a 3 mM. Este bajo incremento explica por qué sólo una minoría de los pacientes son diabéticos según los criterios de la Organización Mundial de la Salud (OMS) (glucemia a las 2 h en la PTOG >= 11 mM) y la mayoría presentan cifras anormales de glucemia en ayunas y/o intolerancia a la glucosa1,25. Los estudios de corte transversal indican que la hiperglucemia de MODY 2 permanece estable o sólo aumenta moderadamente con la edad25,28. Sin embargo, algunos pacientes presentan hiperglucemia progresivamente creciente, aunque al parecer esto es excepcional.
En concordancia con la ligera hiperglucemia hallada en estos pacientes, existe una prevalencia más baja de complicaciones microvasculares de la diabetes (retinopatía y proteinuria) en MODY 2 que en otros subtipos de MODY y en la diabetes tipo 21,29. Además, la conocida asociación de la diabetes tipo 2 o la intolerancia a la glucosa con una serie de factores de riesgo para la enfermedad macrovascular, como la hipertensión, la obesidad y la dislipemia, es muy rara en los sujetos con MODY 2, lo cual concuerda con la baja frecuencia de cardiopatías coronarias en estos sujetos1.
Mutaciones de la glucocinasa, embarazo y crecimiento fetal
La ligera hiperglucemia asintomática asociada con las mutaciones de la glucocinasa puede pasar fácilmente inadvertida en el niño y el adulto joven. Así pues, el diagnóstico de MODY 2 se realiza a menudo durante el embarazo. En algunas series se hallan mutaciones de la glucocinasa en el 1-4% de las mujeres diagnosticadas de diabetes gestacional30,31. Se ha observado que los criterios específicos que inclinan al diagnóstico de una mutación de la glucocinasa en las mujeres que se presentan con diabetes gestacional son la hiperglucemia persistente en ayunas, un pequeño aumento en la PTOG (< 3 mM) y los antecedentes familiares de ligera hiperglucemia30.
Dado que las mutaciones de la glucocinasa ocasionan una ligera hiperglucemia durante todo el embarazo, los recién nacidos suelen ser de peso elevado para la edad de gestación32. Sin embargo, el crecimiento fetal depende también de la situación de la mutación en el propio feto32,33. Si el feto hereda la mutación, disminuye el peso de nacimiento32,33. Ello se debe a que la menor percepción de la glucemia materna por parte del páncreas fetal da lugar a que disminuya la secreción insulínica del feto y, por lo tanto, a un menor crecimiento intrauterino. Así pues, la evolución del embarazo depende de un efecto aditivo: por un lado, la mutación materna, que aumenta el peso fetal; por otro, la mutación fetal, que lo disminuye. Si la madre y el feto presentan una mutación de la glucocinasa, los 2 efectos opuestos se anulan mutuamente y el recién nacido tiene un peso normal32,33. Aunque el peso de nacimiento sea bajo, no se observan diferencias en la talla, el peso o el índice de masa corporal en los sujetos con MODY 2 preadolescentes, adolescentes o adultos, en comparación con sus hermanos no afectados33.
Tratamiento de MODY 2
El tratamiento farmacológico de la hiperglucemia raras veces es necesario en los sujetos con MODY 2, ya que se hallan asintomáticos, la cifra de hemoglobina glucosilada se halla cerca del límite superior de la normalidad y el riesgo de complicaciones es muy bajo. En algunas series, hasta un 2-30% de los pacientes se tratan con hipoglucemiantes orales, o incluso con insulina. Sin embargo, es probable que en la mayoría de estos casos el tratamiento no altere el control glucémico. La secreción de insulina se regula con un aumento del umbral glucémico en los sujetos con MODY 220,21. Si se administra insulina a estos sujetos, probablemente disminuya la secreción insulínica endógena, a menos que se dé una dosis total de reemplazamiento. El papel de los hipoglucemiantes orales es también limitado. Los sujetos con MODY 2 presentan además un mayor umbral para la contrarregulación hipoglucémica34, lo que dificulta considerablemente la reducción crónica de la glucemia. Por lo tanto, una alimentación sana normal probablemente sea el tratamiento más apropiado para la hiperglucemia en los sujetos con MODY 2. Es probable que sólo un porcentaje muy escaso de pacientes con MODY 2 precisen alguna vez tratamiento farmacológico. Cabría conjeturar que estos sujetos también son portadores de otros alelos de susceptibilidad a la diabetes, frecuentes en la población general. Las sulfonilureas estimulan la secreción de insulina por una vía que cortocircuita el metabolismo de la glucosa dependiente de la glucocinasa en las células ß y, por lo tanto, podrían ser útiles para estos pacientes. En los casos raros con importante hiperglucemia en ayunas, la metformina podría ser también útil, aunque el peso corporal sea normal, dado que disminuye la producción hepática de glucosa. No se han valorado las indicaciones y los efectos beneficiosos de estos tratamientos. No es necesario que el seguimiento de los pacientes con MODY 2 sea tan intensivo como en otras formas de diabetes; probablemente es apropiado realizar una revisión anual con determinación de la hemoglobina glucosilada.
Las mujeres con mutaciones de la glucocinasa se tratan a menudo con insulina durante el embarazo, para intentar corregir la hiperglucemia en ayunas y prevenir la macrosomía. Aunque los datos de la bibliografía no son abundantes, se han descrito algunos casos en que el niño ha heredado la mutación y el tratamiento insulínico intensivo de la madre ha ocasionado un bajo peso de nacimiento35. Por lo tanto, debe tenerse en cuenta que tanto la hiperglucemia materna como el fenotipo fetal influyen en la evolución del embarazo32,33. Cabe señalar de nuevo que deben valorarse adecuadamente las indicaciones y los beneficios del tratamiento con insulina en el embarazo de las mujeres con MODY 2.
MODY 3
Las mutaciones heterocigóticas en el gen que codifica el factor de transcripción HNF-1* causan MODY 33, y se han detectado más de 150 mutaciones diferentes en pacientes de diversas procedencias étnicas36 (Bellanné-Chantelot et al, resultados no publicados). Las pruebas sistemáticas para detectar las mutaciones HNF-1* en el United Kingdom Prospective Diabetes Study (UKPDS) mostraron que MODY 3 era responsable de cerca del 3% de los casos en los pacientes con un diagnóstico clínico de diabetes tipo 237.
Fisiopatología de las mutaciones HNF-1*
HNF-1* se expresa en numerosos órganos, como el páncreas, el hígado, el riñón y el intestino. La molécula HNF-1* comprende 3 dominios principales: un dominio N-terminal, que interviene en la dimerización de las proteínas; un dominio de ligazón con el ADN, y un dominio C-terminal, que interviene en la transactivación. La dimerización, como homodímero o heterodímero con HNF-1ß, es necesaria para la transactivación de los genes destinatarios. Las mutaciones que causan MODY 3 se han hallado en los 3 dominios funcionales, y se ha identificado un lugar de riesgo mutacional en el exón 438. Los estudios funcionales realizados con varias de estas mutaciones han confirmado que existe una disminución de las propiedades de ligazón al ADN o una pérdida de la transactivación39-41. Además, se han descrito variantes del promotor que disminuyen su actividad transcripcional y cosegregan con la diabetes42. Sin embargo, no se observó una clara relación genotipo-fenotipo entre el tipo o la localización de las mutaciones y la expresión variable de la enfermedad43,44.
Aunque no se conoce bien la fisiopatología de la hiperglucemia de MODY 3, probablemente intervienen la señalización glucolítica defectuosa y una producción mitocondrial anormal de la adenosintrifosfato en las células ß pancreáticas45,46. Además, se ha observado que HNF-1* desempeña un papel en la transcripción de numerosos genes que intervienen en el desarrollo y la función de las células ß pancreáticas, incluido el gen de la insulina47-49. La secreción de insulina en respuesta a la glucosa, tanto en la primera como en la segunda fases, y en respuesta a la arginina, está notablemente reducida en los pacientes con MODY 350,51. Estos defectos ya pueden detectarse en sujetos jóvenes normoglucémicos portadores de las mutaciones51. En los sujetos con MODY 3 se observa también una menor estimulación para la utilización y oxidación de la glucosa y para la distribución no oxidativa de la glucosa, así como una menor supresión de la producción endógena de glucosa50-52. Sin embargo, se ha hallado que estos fenómenos son esencialmente secundarios a la insulinopenia ocasionada por el defecto de las células ß en los sujetos diabéticos52, y no se ha observado resistencia a la insulina en los sujetos normoglucémicos portadores de la mutación51.
Características clínicas de MODY 3
La presentación clínica de MODY 3 difiere de la de MODY 2 en varios aspectos, especialmente en cuanto a la gravedad y al curso natural del defecto de secreción de insulina y a la hiperglucemia asociada. En 2 series de pacientes MODY 37,50, la glucosa plasmática en ayunas y la determinada a las 2 h de la PTOG eran más elevadas que en los pacientes con MODY 27. En un estudio multicéntrico realizado en Europa25, la cifra de glucosa plasmática en ayunas era similar en los pacientes con MODY 3 y con MODY 2, pero la cifra a las 2 h era mayor en los primeros (media ± desviación estándar: 15,5 ± 3,6 frente a 9,0 ± 1,8 mM). Sin embargo, en dicho estudio no se incluyó a los pacientes tratados con insulina, es decir, aquéllos con hiperglucemia grave. En la PTOG, el aumento a las 2 h en relación con la cifra en ayunas era superior a 3 mM en cerca del 70% de los pacientes con MODY 3, mientras que el citado aumento se hallaba sólo en menos del 30% de los sujetos con MODY 225. El deterioro con la edad en las cifras de glucosa plasmática en ayunas, y especialmente a las 2 h, es importante en los sujetos con MODY 3; en cambio, es muy ligero en los que presentan MODY 225,53.
La expresión clínica de MODY 3 es muy variable entre las distintas familias, así como dentro de una misma familia. En la mayoría de los casos, el comienzo clínico de la diabetes ocurre después de la pubertad, a una edad media de 22 a 26 años. Sin embargo, la penetrancia no es completa y los factores ambientales y otros de carácter genético modulan la expresión clínica. Los portadores de la mutación pueden ser normoglucémicos, mientras que sus hermanos son muy hiperglucémicos, o lo fueron a edades análogas. En una familia, 2 portadores de la mutación se hallaban todavía normoglucémicos a los 46 y 87 años de edad54. Los hermanos normoglucémicos portadores de una mutación presentan una sensibilidad normal a la insulina, pero su secreción insulínica es anormal51. Estos sujetos tienen riesgo de desarrollar diabetes si se produce resistencia a la insulina, por ejemplo, durante el embarazo o al aumentar de peso. En el estudio de Letho et al50, el 38% de las mujeres con MODY 3 habían presentado diabetes gestacional. En dicho estudio, los portadores no diabéticos eran más delgados (índice de masa corporal medio: 21 kg/m2) que los diabéticos (índice de masa corporal: 25 kg/m2)50. Además, 2 estudios han mostrado que la edad de diagnóstico de la diabetes en los descendientes portadores de HNF-1* es 5-10 años menor cuando se diagnosticó la diabetes materna antes del embarazo, en comparación con los casos en que se diagnosticó después44,55. Estas observaciones indican que la exposición intrauterina a la hiperglucemia materna modula la penetrancia de las mutaciones HNF-1*. Además, se ha señalado que la edad de comienzo de la diabetes es hereditaria en las familias con MODY 3 y se han establecido mapas genéticos putativos de los locus modificadores56.
Los síntomas iniciales pueden variar asimismo entre o dentro de las familias. La mayoría de los sujetos tienen glucosuria y poliuria importantes en el momento del diagnóstico, lo que probablemente limita la intensidad de la hiperglucemia. De hecho, en los sujetos con MODY 3 se ha descrito un menor umbral para la glucosuria y un descenso del 50% en la reabsorción tubular de glucosa57,58. Los estudios realizados en ratones knock-out HNF-1* han mostrado que este defecto se debe a una menor expresión tubular proximal del cotransportador sodio-glucosa, de baja afinidad y alta capacidad, que se halla bajo el control transcripcional de HNF-1*58. Esta anomalía renal puede observarse en portadores normoglucémicos de mutaciones en HNF-1*58 y puede ser responsable de la antigua observación de que la glucosuria precede a la diabetes. Los pacientes con MODY 3 tienen habitualmente un peso normal y no presentan rasgos metabólicos asociados con la diabetes tipo 2 (síndrome metabólico). Así pues, el diagnóstico de MODY 3 puede sospecharse clínicamente en pacientes jóvenes con antecedentes familiares indicativos e hiperglucemia importante, aunque con escasa o nula cetosis. Sin embargo, ninguno de estos criterios posee un valor absoluto. En el estudio UKPDS no siempre había antecedentes familiares conocidos en los pacientes con mutaciones en HNF-1*37. Se han descrito algunos casos en que la diabetes se reveló a través de una verdadera cetoacidosis. De hecho, en los sujetos jóvenes, la intensidad de la hiperglucemia y de los síntomas clínicos puede conducir al diagnóstico de diabetes tipo 1. En este contexto, es importante investigar los marcadores inmunológicos (autoanticuerpos antiácido glutámico descarboxilasa y antitirosinfosfatasa) e inmunogénicos (haplotipos del complejo principal de histocompatibilidad de susceptibilidad de clase II DR3 y DR4) de la diabetes tipo 1. En el 5-10% de los pacientes que no poseen estos marcadores puede hallarse una mutación de HNF-1*. En un estudio realizado en Dinamarca entre 39 pacientes considerados inicialmente como de tipo 1 según los criterios de la OMS, pero sin haplotipos del complejo principal de histocompatibilidad de susceptibilidad de clase II, y con antecedentes familiares de diabetes, 4 presentaban una mutación de HNF-1* que cosegregaba con la diabetes59. De modo similar, entre 28 pacientes japoneses diagnosticados de diabetes tipo 1, pero sin autoanticuerpos antiinsulares, 2 de ellos presentaban una mutación en HNF-1*60. Así pues, algunos sujetos con la denominada "diabetes tipo 1 no autoinmunitaria", según la nueva clasificación de los síndromes diabéticos, podrían presentar MODY 3.
Las complicaciones microvasculares son frecuentes en los pacientes con MODY 3. Tras 16 años de diabetes, la prevalencia de retinopatía se halla en torno al 50%, y las formas graves de esta enfermedad (preproliferativa, proliferativa, edema macular, necesidad de fotocoagulación con láser) afectan al 15-20% de los pacientes29,61,62. La nefropatía incipiente (microalbuminuria patológica) está presente en cerca del 20% de los casos. Por lo tanto, al considerar la duración de la diabetes y el nivel del control glucémico, la prevalencia de las complicaciones microangiopáticas es la misma que en la diabetes tipo 1 y tipo 262. En cambio, la frecuencia de hipertensión, concentraciones anormales de lípidos y macroangiopatía (cardiopatía coronaria) es menor que en la diabetes tipo 229,37,62.
Se ha descrito la aparición de adenomatosis hepática por inactivación bialélica de HNF-1*, ya sea por un fenómeno somático doble o por un solo fenómeno en asociación con una mutación en la línea germinal de HNF-1*63. Esta observación induce a pensar que HNF-1*puede funcionar como supresor tumoral. La adenomatosis hepática es una rara enfermedad definida por la presencia de numerosos adenomas en un parénquima hepático normal. La enfermedad puede permanecer clínicamente muda y sólo detectable por ecografía, o bien puede ocasionar hemorragias importantes. La adenomatosis hepática se ha descrito recientemente en familias con MODY 364. Se desconoce su prevalencia en portadores de MODY 3, pero su potencial gravedad suscita la cuestión de realizar pruebas para su detección clínica en todos los portadores de mutaciones en HNF-1*. Es necesario realizar nuevos estudios para valorar si un espectro mutacional específico de HNF-1* se asocia con la adenomatosis hepática.
Tratamiento de MODY 3
Existe una variación considerable en la gravedad de la hiperglucemia de MODY 3, que se refleja en el tratamiento de la diabetes. En algunas series, hasta una tercera parte de los pacientes se controlan tan sólo con dieta7,50,61. Cuando es necesario un tratamiento farmacológico, la utilización de sulfonilureas parece una elección lógica, pues se ha descrito una alta sensibilidad de los pacientes con MODY 3 a estos fármacos65,66. Recientemente se ha publicado un ensayo farmacológico cruzado de distribución aleatoria sobre el empleo de glicazida y metformina en pacientes con MODY 3 o diabetes tipo 2, emparejados en cuanto al índice de masa corporal y al grado de hiperglucemia66. En los sujetos con MODY 3, la respuesta a la glicazida fue 5 veces mayor que a la metformina en términos de reducción de la glucosa plasmática en ayunas. La respuesta a la glicazida fue 4 veces mayor en los sujetos con MODY 3 que en los afectados de diabetes tipo 2. Además, en dicho estudio los pacientes con MODY 3 conservaban bien la respuesta secretoria de insulina frente a la tolbutamida intravenosa, a pesar de existir una respuesta muy disminuida frente a la glucosa intravenosa66. Esta sensibilidad a las sulfonilureas puede observarse incluso en los sujetos con MODY 3 que presentan grave descompensación diabética. Nosotros hemos observado el caso de una mujer joven con glucemia en ayunas de 27 mM y hemoglobina glucosilada del 15,9% cuyo control glucémico se normalizó (hemoglobina glucosilada del 6,1%) con 15 mg/día de gliburida67.
En algunas series, hasta un 17-47% de los pacientes con MODY 3 se tratan con insulina7,50,61,68, y éstos tienen más edad que los tratados con hipoglucemiantes orales o dieta68. Esto podría estar relacionado con un declive progresivo de la secreción insulínica53. Sin embargo, se desconocen la frecuencia y la tasa reales del fallo secundario del tratamiento con sulfonilurea en los pacientes con MODY 3. En un estudio no se observó deterioro del control glucémico en los sujetos con MODY 3 después de sustiuir el tratamiento insulínico por la sulfonilurea69. Todas estas observaciones llevan a pensar que podría ser útil valorar la respuesta a las sulfonilureas en todos los sujetos con MODY 3 que precisan tratamiento farmacológico para la hiperglucemia. No obstante, en el embarazo, las mujeres con MODY 3 que necesitan tratamiento farmacológico de la hiperglucemia deben cambiar a insulina.
Debido a las características clínicas de MODY 3 (gravedad y progresión de la hiperglucemia, edad de comienzo temprana, con la consiguiente larga duración de la diabetes, frecuencia de las complicaciones microvasculares), proponemos que los pacientes con MODY 3 deben someterse a controles clínicos frecuentes. Debe ofrecerse la investigación familiar de la mutación, especialmente en las mujeres en edad fértil, para evitar los riesgos teratogénicos inherentes a la hiperglucemia no diagnosticada durante las primeras semanas de la gestación. Los familiares no diabéticos portadores de la mutación deben investigarse una vez al año, o al menos cada 2 años, en busca de hiperglucemia. Puede ser necesario practicar una PTOG, pues se ha demostrado que la medición de la glucosa plasmática en ayunas tiene una escasa sensibilidad para diagnosticar MODY 325. Como estos sujetos tienen el riesgo de desarrollar diabetes, especialmente si existe resistencia a la insulina, deben alentarse los hábitos de comida sana y ejercicio regular para evitar el sobrepeso.
MODY 1
MODY 1 es un raro subtipo de diabetes producido por mutaciones en el gen que codifica a HNF-4*2, un receptor nuclear huérfano. Se conocen sólo en parte los mecanismos moleculares en virtud de los cuales una reducción de la actividad transcripcional de HNF-4* da lugar a defectos en las células ß y diabetes. La pérdida funcional de HNF-4* altera la expresión de los genes que intervienen en el transporte de glucosa y en la producción de adenosintrifosfato70. HNF-4* es asimismo un regulador anterógrado de la expresión de HNF-1*(MODY 3). Por lo tanto, la menor expresión de HNF-1* a consecuencia de una mutación en HNF-4* podría contribuir al fenotipo MODY 1. Esta hipótesis viene avalada por la observación de que una mutación en el sitio de unión de HNF-4* en el promotor del gen de HNF-1* da lugar a MODY 371. Además, un segundo promotor anterógrado a distancia en el gen que codifica a HNF-4*contiene sitios de unión para HNF-1*, HNF-1ß e IPF-1. Se ha hallado que una mutación localizada en un sitio de unión para IPF-1 cosegregaba con la diabetes en una familia con MODY 172. Estos resultados apuntan a la existencia de una red que conecta diversos factores de transcripción que intervienen en el desarrollo y la función de las células ß pancreáticas.
La mayor parte de la información clínica disponible para MODY 1 proviene del seguimiento prospectivo durante 40 años de la gran genealogía RW73,74. Los individuos afectados suelen presentar una forma grave de diabetes que se asocia a menudo con complicaciones microvasculares. Los miembros de la familia suelen ser delgados y la diabetes aparece a diversas edades, entre los 7 y 40 años, pero a la larga se produce hiperglucemia en ayunas en el 95% de los portadores de la mutación. Así pues, la penetrancia es casi completa, pero la expresión clínica es variable. Un descenso progresivo de la secreción endógena de insulina es la causa de la disminución de la respuesta a las sulfonilureas, en una cuantía del 1-4% anual. El 30-40% de los pacientes no responden a las sulfonilureas después de 3 a 25 años de hiperglucemia, y la diabetes adquiere semejanza con la diabetes tipo 1 "lábil". A la inversa, en algunos pacientes se ha observado una buena respuesta al tratamiento con sulfonilureas durante períodos de hasta 40 años. Se desconoce si estas características clínicas son aplicables a los pacientes de otras genealogías MODY 1, dado el limitado número de familias descritas hasta el momento.
MODY 5
MODY 5 se describió inicialmente como la asociación de diabetes de comienzo temprano y nefropatía, causada por mutaciones en el gen que codifica a HNF-1ß5. En observaciones clínicas aisladas se describieron luego fenotipos heterogéneos, tales como diabetes clínicamente similar a MODY 3, anomalías renales, malformaciones genitales y trastornos de la función hepática75-89. Hasta el momento, los fenotipos de todas las familias con MODY 5, a excepción de 278,87, eran compatibles con la herencia autosómica dominante de mutaciones independientes.
MODY 5 parece más frecuente de lo que cabría esperar por el escaso número de casos descritos en la bibliografía. Nosotros hemos descrito recientemente una serie de 13 pacientes de 8 familias con MODY 5, con diferentes mutaciones localizadas en el dominio de unión con el ADN, incluidos 5 cambios de sentido (mutaciones missense)90. Se halló una nueva mutación en 2 familias. Existía diabetes en 10 de los 13 portadores de mutación (77%). La diabetes era clínicamente manifiesta en 5 sujetos, incluidos 2 pacientes jóvenes en los que apareció de forma aguda a las edades de 1 y 13 años. En otros 5 pacientes, la diabetes se halló en las pruebas de detección a las edades de 19 a 38 años. Todos los sujetos eran delgados. Las concentraciones de péptido C en respuesta al glucagón intravenoso mostraron grados variables de déficit en la secreción insulínica (límites: 0,23 a 1,95 nM; media ± desviación estándar: 0,95 ± 0,63 nM; en comparación, era de 2,30 ± 0,48 nM en 10 controles no diabéticos, y de 0,42 ± 0,28 nM en 90 pacientes con diabetes tipo 1 de comienzo reciente). Seis sujetos se trataron con insulina. Un hallazgo llamativo fue la presencia de atrofia pancreática, detectada mediante tomografía computarizada en 5 de los 6 sujetos (83%) investigados. Se observó déficit subclínico del páncreas exocrino en 6 de 7 sujetos (86%). En 9 pacientes de 18 a 41 años se halló afectación renal, con cambios morfológicos (disminución del tamaño renal, quistes renales, anomalías pelvicaliciales) e insuficiencia renal lentamente progresiva. Se hallaron quistes glomerulares en muestras renales de 2 pacientes, y oligomeganefronia en uno. Por ecografía fetal se observaron riñones displásicos en 3 casos que se presentaron con insuficiencia renal neonatal transitoria. Existían anomalías del tracto genital en una mujer (útero bicorne) y en 4 varones (quistes del epidídimo, infertilidad por azoospermia y agenesia bilateral del conducto deferente) entre los casos índice. Se observó aumento de los títulos de enzimas hepáticas en 11 de 13 pacientes (85%), aunque sin acompañarse de síntomas clínicos. Estos diversos fenotipos recuerdan a los previamente descritos en asociación con mutaciones en HNF-1ß, incluidos los riñones displásicos descubiertos por ecografía fetal78,80-82, los quistes renales asociados con diabetes en pacientes adultos80,91, la enfermedad glomeruloquística renal familiar80,82,87 y la oligomeganefronia77. El perfil clínico de MODY 5 encaja bien con el patrón de expresión de HNF-1ßdurante los primeros estadios de la organogénesis de los tractos urinario y genital, el hígado y las vías biliares y el páncreas, como se ha descrito en el ratón92-94 y en el ser humano81.
Dos estudios recientes han arrojado luz sobre la fisiopatología de los trastornos renales asociados con MODY 595,96. Se ha observado que el promotor del gen murino Pkhd1 contiene un sitio de unión con HNF-1, y que HNF-1ß y HNF-1* se unen específicamente al promotor Pkhd1 y estimulan la transcripción del gen95,96. Ratones transgénicos con inactivación renal específica de HNF-1ß95 o con expresión de un HNF-1ß96 mutante bajo el control de un promotor renoespecífico desarrollaron enfermedad poliquística renal. Las transcripciones Pkhd1 se hallaban ausentes en las células que tapizaban los quistes, pero estaban presentes en los túbulos circundantes normales96. Se observó que el gen Pkhd1 se expresaba a concentraciones elevadas en el riñón fetal y adulto, y a concentraciones inferiores en el hígado, páncreas, corazón, estómago, intestino, músculo esquelético, útero, testículo y placenta. Pkhd1 codifica la poliductina (o fibrocistina), una proteína de la membrana de 450 kD, que se expresa en el asa de Henle, en los túbulos colectores renales y en los conductos biliares intrahepáticos. También se halló expresión en los hepatocitos y los conductos pancreáticos, así como en el epidídimo y testículo en desarrollo. En el ser humano, las mutaciones en Pkhd1 causan la enfermedad poliquística renal autosómica recesiva97, un trastorno quístico con asociación de quistes en los conductos colectores renales que conducen a insuficiencia renal en lactantes y niños, y disgenesia biliar, que produce unos conductos biliares intrahepáticos aberrantes y posteriormente fibrosis portal.
Se sabe actualmente que MODY 5 abarca un amplio espectro clínico, variable entre las familias y dentro de una misma familia. Así pues, está justificado realizar pruebas de detección para las mutaciones en HNF-1ß en sujetos diabéticos no obesos con nefropatía no diabética lentamente progresiva, sobre todo cuando existen malformaciones renales o genitales o trastornos funcionales hepáticos, sin tener en cuenta los antecedentes familiares de diabetes.
OTROS TIPOS DE MODY
IPF-1 consituye un elemento crítico para el desarrollo embrionario de los islotes pancreáticos, así como para la regulación transcripcional de los genes histoespecíficos del páncreas endocrino, como los de insulina, transportador 2 de la glucosa y glucocinasa en las células ß, y el gen de la somatostatina en las células *. IPF-1 se expresa en todas las células del mamelón pancreático, y su ausencia en el ratón detiene el desarrollo en el estadio de mamelón, lo que conduce a la agenesia pancreática98. MODY 4, por mutaciones en el gen que codifica a IPF-1, es muy raro. Se ha hallado una deleción que cosegrega con MODY en una familia numerosa con vínculos consanguíneos4. Esta mutación da lugar a un codón stop prematuro y a una proteína desprovista de un dominio crucial para la unión con el ADN. El fenotipo de los sujetos heterocigóticos para la mutación oscilaba desde la normalidad hasta la diabetes no insulinodependiente, pasando por la intolerancia a la glucosa. Un niño homocigótico para la mutación nació con agenesia pancreática y presenta diabetes e insuficiencia exocrina99.
El factor de transcripción Neuro-D1 interviene en la regulación del desarrollo del páncreas endocrino. En ratones homocigóticos para un trastorno de los objetivos de Neuro-D1, la morfogenia de los islotes pancreáticos es anormal y desarrollan hiperglucemia, debido en parte a una expresión insuficiente del gen de la insulina100. Se observó que las mutaciones en Neuro-D1 cosegregaban con la diabetes de comienzo temprano y con una transmisión semejante a la autosómica dominante en 2 familias, las únicas MODY 6 descritas hasta la fecha6.
CONCLUSIONES
El establecimiento del diagnóstico clínico de MODY y del diagnóstico molecular de los subtipos de MODY es importante para una asistencia apropiada del paciente. Los pacientes con mutaciones de la glucocinasa, HNF-1* o HNF-1ß presentan cursos clínicos diferentes en cuanto a la progresión de la hiperglucemia, las necesidades de tratamiento, el riesgo de complicaciones y la evolución del embarazo. Las pruebas moleculares de detección para los genes relacionados con MODY se hallan disponibles como método diagnóstico sistemático en diversos países europeos. Dado que estas pruebas son todavía caras y laboriosas, debe seleccionarse cuidadosamente a los pacientes candidatos a ellas. Sin embargo, es importante señalar que debe sospecharse el diagnóstico de MODY en una gama de situaciones clínicas más amplias de lo que inicialmente se había descrito. Además, la falta de antecedentes familiares de diabetes no es un criterio de exclusión absoluto, pues a menudo no se han realizado pruebas para detectar la diabetes en los progenitores, y una ligera hiperglucemia en ayunas puede pasar inadvertida durante varias décadas. Es necesario realizar nuevas investigaciones para definir con más precisión las características fenotípicas y los marcadores clínicos que faciliten el hallazgo de un diagnóstico molecular positivo para los diversos subtipos de MODY.
